

1. 서 론
도전 페이스트(paste)의 가격 경쟁력 향상을 위해 기존 에 사용되던 순수 Ag 필러(filler) 소재를 대신하여 Ag 코 팅 Cu 필러 소재를 적용하고자 하는 연구가 최근 들어 활발히 진행되고 있다.1-11) 즉, 이는 Cu는 Ag 다음으로 전 기전도도 및 열전도도가 우수한 금속이면서 가격 경쟁 력이 월등히 우수하므로 필러 입자 소재의 코어(core) 금 속으로 사용하고, 이 Cu 입자 표면을 Ag로 코팅하여 Cu 와 달리 우수한 내산화성을 가지는 필러를 제조하여 사 용하고자 하는 연구이다.
Ag 코팅 Cu 필러 소재를 적용한 도전 페이스트의 특 성과 경쟁력은 다음 두 종류의 선행보고를 통해 확인된 다. Cu 등은 열 개시제(thermal initiator)를 함유하는 비 닐에스테르 레진(vinyl ester resin) 기반 도전 페이스트 의 제조에서 필러 소재로 기존 순수 Ag 분말에 Ag 코 팅 Cu 분말을 13~23 %까지 섞어 사용함으로써 순수 Ag 분말만을 사용한 경우와 유사한 전기전도도를 얻을 수 있었다.11) 즉, 이 경우에서는 Ag 코팅 Cu 분말의 사용 양만큼 가격 경쟁력을 확보하게 된다. 또한 Eom 등은 도전성 페이스트의 필러 소재로서 솔더 분말과 Cu 분 말을 혼합하여 사용한 하이브리드 페이스트의 우수한 특 성들을 보고하였는데,12) 이 페이스트에서 순수 Cu 분말 대신 Ag 코팅 Cu 분말을 사용한 경우 보다 우수한 전 기전도도를 얻을 수 있었다. 그러나 Ag 코팅 Cu 필러만 을 사용한 도전 페이스트의 경화 후 전기전도도는 순수 Ag 필러를 사용한 경우에 비해 1 order, 즉 10배 이상 높은 것으로 보고되고 있는데,8,10) Ag 코팅층의 dewetting 현상이 일어나지 않는 200 °C 미만의 온도에서 경화를 실시하였고, 질소 분위기 중 경화 시에는 순수 Ag 필 러를 사용한 경우의 전기전도도에 버금가는 특성이 획 득되었음을 고려할 때 이는 불완전한 Ag 코팅으로 인 해 경화 중 Cu 코어 입자가 산화되었기 때문으로 분석 된다.10) 따라서 Ag 코팅 Cu 분말을 필러로 사용한 도 전 페이스트에서 Ag 코팅 Cu 필러의 품질, 즉 Ag 코 팅층의 완전한 피복 특성은 향후 페이스트의 도전 특성 에 결정적인 영향을 미치는 매우 중요한 인자가 된다. 그리고 Ag 코팅층의 제조가 습식 공정으로 이루어지고 있음을 고려할 때 Cu 입자 표면의 산화층을 제거하는 전처리 공정은 이후 환원반응으로 도금되는 Ag 코팅층 의 품질, 즉 피복 정도를 결정하는 가장 중요한 공정으 로 고려되고 있다.9) 또한 Cu 입자가 담겨있는 전처리 용 액에 Ag 도금액을 투입하는 일반적인 코팅법에서는 전 처리 용액의 농도가 높을수록 Ag의 도금동안 Cu 입자 들에 홀(hall)이 생성되는 거동이 더욱 심하게 관찰되므 로 전처리 용액의 농도는 최대한 진하지 않게 적정한 농 도로 사용하는 것이 바람직하다.9) 따라서 Ag 코팅 Cu 필러의 품질 향상을 위해서는 Cu 입자 표면의 산화층 을 제거하는 전처리 공정의 최적화와 이를 기반으로 한 후속 Ag 도금의 품질 향상이 요청된다.
Ag 코팅 Cu 입자의 제조 공정에서 몇몇 주요 공정변 수의 영향이 보고된 바 있고,4,6) 이 소재가 현재 국내외 에서 양산, 판매되고 있는 상황임에도 불구하고, Cu 입 자 표면을 Ag로 도금시키기 전에 Cu 입자 표면의 산 화층을 안정적으로 제거하기 위한 연구결과 및 전처리 공정의 영향에 대한 연구결과 보고는 매우 부족한 상황 이다. 따라서 본 연구에서는 Cu 입자 표면 산화층의 제 거를 위해 가장 일반적으로 사용되고 있는 전처리용액 인 ammonium hydroxide/ammonium sulfate 혼합용액의 농도 변화에 따른 Cu 입자상에서의 산화층 제거 특성 변화와 이에 따른 후속 Ag 도금의 품질 변화에 대한 실 험과 논의를 실시하고자 한다.
2. 실험 방법
2.1 동분말의 전처리
동분말(> 99.6 %, mean particle size: 2 μm, JoinM) 입자 표면의 산화층을 제거하기 위하여 100 ml의 증류수 에 ammonium hydroxide(NH4OH, 28-30 % NH3 basis, Sigma-Aldrich)를 혼합한 제 1용액과, 또다른 100 ml의 증류수에 ammonium sulfate[(NH4)2SO4, 99 %, Sigma- Aldrich]를 용해시킨 제 2용액을 각각 준비한 후 두 용 액을 혼합한 암모늄 기반 전처리용액을 사용하였다. 제 1용액과 제 2용액은 다양한 농도로 제조되어 사용되었 고, 이후 이 혼합 용액에 1 g의 표면 산화된 동분말을 장 입하여 특정 시간동안 교반시킨 후 장입된 동분말을 회 수하였다. 100 ml의 증류수에 0.788 ml의 NH4OH를 혼합 한 제 1용액과 100 ml의 증류수에 2.6428 g의 (NH4)2SO4 를 용해시킨 제 2용액을 제조하여 이들을 혼합한 다음 산화된 동분말 1 g을 이 혼합 용액에 장입, 교반하여 전 처리를 실시하였을 경우 이 NH4OH와 (NH4)2SO4의 첨 가량은 농도 X1이라 명명하였고, 동일 증류수 양에 NH4OH와 (NH4)2SO4를 각각 2배 및 3배 용해시킨 경우 는 농도 X2 및 X3로 언급하였다.
분말 회수를 위해 교반을 중지시킨 후 1분 이상 분말 을 가라앉혀준 다음 상층액을 제거하였다. 상층액 제거 후 메탄올을 다시 장입하여 7000 rpm의 속도에서 원심 분리하는 과정을 3회 반복하여 용액 중 불순물을 충분 히 제거한 다음 최종적으로 미량의 메탄올이 함유된 분 말 슬러리를 진공 챔버에서 상온 건조하여 전처리된 분 말을 제조하였다.
전처리 조건에 따른 분말의 상분석은 X선 회절분석기 (X-ray diffractometer, XRD, X'pert PRO-MPD, PANalytical) 를 사용하여 실시하였다. 건조된 전처리 분말은 질소 팩 킹하여 보관하다 XRD 측정 직전에 짧은 시간동안만 대 기 중에 노출시켰다.
2.2 동판의 전처리
Auger 전자분광법(Auger electron spectroscopy, AES) 을 통한 산화층의 두께 변화를 측정하기 위해 동판을 사 용한 산화 실험과 전처리를 통한 산화층 제거 실험을 실 시하였는데, 사용된 동판(> 3N)의 크기는 10 × 10 mm였 다. 전처리를 위해서 앞서와 동일한 방법으로 준비된 다 양한 농도의 암모늄 기반 혼합 전처리용액에 동판을 5 분간 담구었으며, 이후 꺼내어 메탄올 내에서 초음파 처 리로 세척하고 진공 건조하여 전처리된 동판 시편을 준 비하였다.
전처리된 동판은 Auger 전자분광법(Auger electron spectroscopy, AES, PHI 700Xi, Physical Electronics, Inc.)을 사용하여 표면으로부터 깊이에 따른 원소량 분포 변화를 측정하였다. 전처리 동판은 질소 팩킹하여 보관 하다 AES 측정 직전에 짧은 시간동안만 대기 중에 노 출시켰다.
2.3 동분말의 은도금
제조된 200 ml의 전처리용액에 1 g의 동분말을 장입한 후 5분간 교반하며 전처리한 다음 화학도금을 위한 환 원제로 0.4 g의 potassium sodium tartrate(C4H4KNaO6, 99 %, Oriental Chemical Industries)를 추가로 용해시킨 혼합용액을 제조하였다. 이후 5 ml ammonium hydroxide 에 0.175 g silver nitrate(AgNO3, 99.8 %, Mw: 169.87, Daejung Chemical & Metals Co., Ltd.)을 용해시킨 도 금용액을 상기 동분말 함유 혼합용액에 1분에 걸쳐 방 울방울 장입한 다음 충분히 반응이 일어나게끔 20분간 유지시켜주었다. 이러한 silver nitrate 장입량은 용해된 Ag 이온들이 100 % 환원될 경우 이론적으로 회수되는 Cu와 Ag의 총 분말 무게 대비 15 %에 해당되는 양이 었다. 상기 혼합용액은 도금용액의 장입 시점에서부터 반 응 완료시까지 자기교반자로 지속적으로 교반시켜 균일 한 도금을 유도하고자 하였다. 반응 완료 후에는 교반을 멈추고 용액 중에 부유중인 분말을 1분 이상 가라앉혀 준 뒤 7000 rpm에서 원심분리를 실시하였다. 이후 상부 용액 제거 후 메탄올을 채워 원심분리하는 과정을 3회 반복 실시한 뒤 최종적으로 상부 용액을 제거하고, 진 공 챔버에서 분말 슬러리의 상온 건조를 실시하였다.
건조된 Ag 코팅 Cu 분말의 표면 미세구조는 주사전 자현미경(scanning electron microscope, SEM, VEGA3, Tescan)으로 관찰하였다.
3. 결과 및 고찰
3.1 동분말 및 동판의 전처리
Fig. 1(a)는 200 °C의 대기 중에서 1시간동안 강제 산 화된 동분말을 농도 X1의 전처리용액으로 전처리 시 전 처리 시간에 따른 XRD 결과이다. 전처리 시간의 변화 에도 불구하고 Cu2O 상 피크(peak)의 세기(intensity) 변 화는 쉽게 확인하기 어려웠으나, 전처리 시간이 증가함 에 따라 Cu 상 피크의 세기는 확연히 증가하였다. XRD 측정이 시료의 표면 부위 위주로 진행됨을 고려할 때 이 와 같은 결과는 전처리 시간의 증가에 따라 Cu2O 표면 산화층이 평균적으로 점차 얇아지고 있음을 의미한다. 그 결과 전처리 시간 변화에 따른 Cu 상 주(main) 피크의 세기 대비 Cu2O 상 주 피크 세기 비의 변화값은 Fig. 1(b)와 같았다. Cu2O 상 주 피크의 상대적인 세기, 즉 표면 산화층의 평균적인 두께는 5분간의 전처리 시간까 지 점차 감소하다가 이후 추가 전처리 시간동안에는 다 소 증가하다 포화되는 거동이 관찰되었다. 이와 같은 결 과는 ammonium hydroxide/ammonium sulfate 혼합 전 처리용액의 Cu2O 표면 산화층 제거 능력을 보여주는 한 편 최적의 전처리 시간이 존재함을 나타낸다.
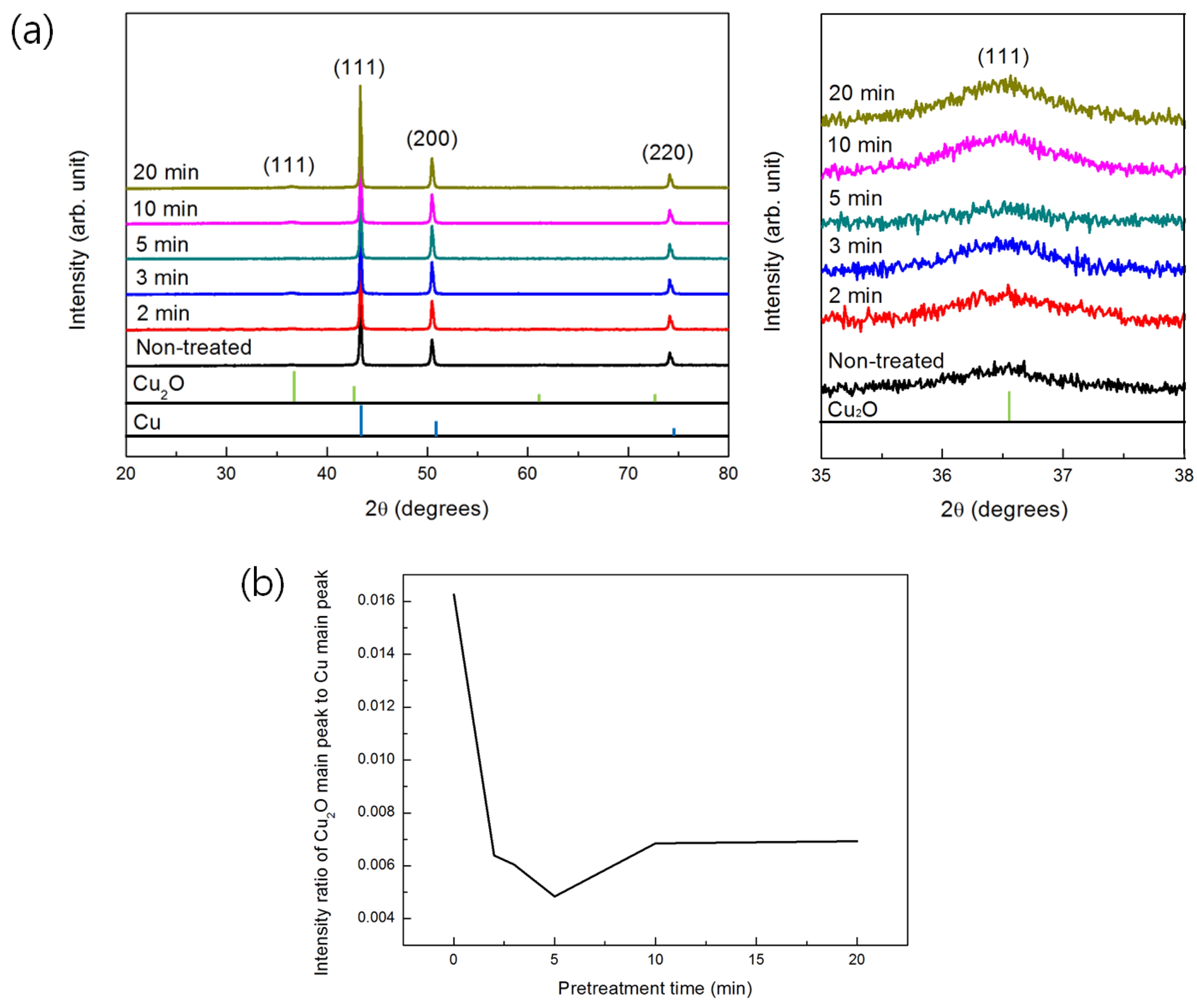
Fig. 1.
(a) XRD patterns and (b) intensity ratios of Cu2O main peak to Cu main peak in the Cu powders pretreated for different times with the solution of X1 concentration after oxidation in air at 200 °C for 1 h.
NH4OH와 (NH4)2SO4 혼합 전처리용액이 Cu2O 산화층 을 제거하는 반응은 다음의 반응식으로 해석된다.
즉, NH4OH와 (NH4)2SO4 혼합으로 인해 암모니아와 황산이 생성되고, 이 암모니아와 황산은 Cu2O 산화층과 반응하여 수용성의 [Cu(NH3)2]2SO4 염을 형성시키므로 전 처리용액의 색상은 푸른빛으로 변화하게 된다. 상기 두 반응을 합치면 아래와 같은 반응식으로 정리될 수 있다.
200 °C의 대기 중에서 1시간동안 강제 산화시킨 동판 을 X1의 전처리용액으로 5분간 전처리한 경우의 AES 결과는 Fig. 2와 같다. Fig. 2의 실선은 강제 산화된 시 편의 Cu 및 O의 농도변화 결과이고, 점선은 전처리 후 Cu 및 O의 농도변화이다. 전처리 전 약 50 nm에 이르 던 산화층(대부분이 Cu2O 상임)의 두께가 전처리 후에 는 약 20 nm까지 감소한 것을 확인할 수 있어 5분 동 안의 산화층 평균 제거속도는 약 6 nm/min에 이르는 것 을 알 수 있었다. 이러한 결과는 X1 혼합 전처리용액 의 산화층 제거 효과를 다시 한번 증빙하는 한편, 약 16 nm 두께의 산화층이 여전히 잔존하는 문제점도 제시하 였다. 당 시편은 대기 중 노출이 극도로 억제되어 자연 발생 산화층(native oxide layer)의 생성 및 성장이 최소 화되었음을 예상할 때 그 잔존 산화층의 대부분은 X1 전처리용액에 의한 불충분한 산화층 제거 결과에 기인 한 것으로 분석되었다.
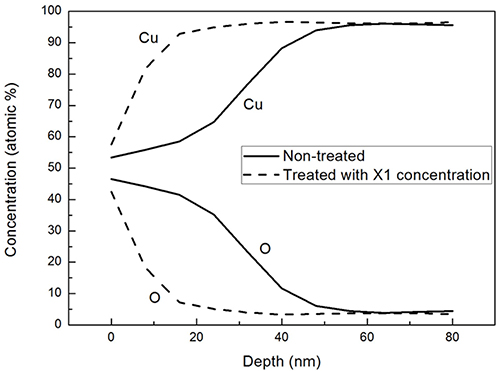
Fig. 2.
AES elemental depth profiles of Cu plates before and after the pretreatment of 5 min with the pretreatment solution of X1 concentration after oxidation in air at 200 °C for 1 h.
이후 동분말 표면의 산화층을 보다 완벽히 제거하기 위 해 전처리용액의 농도를 더욱 증가시킨 후 동일한 전처 리 작업을 실시해 보았다. Fig. 3은 200 °C의 대기 중에 서 1시간동안 강제 산화된 동분말을 5분간 전처리 시 전 처리용액의 농도에 따른 전처리 후 분말의 XRD 결과 이다. 앞서 설명되었듯이 강제 산화 후 확연히 관찰되 었던 Cu2O 상 피크가 X1 전처리용액으로 전처리 시 그 세기가 다소 줄어드는 결과를 관찰할 수 있었고, 이후 전처리용액의 농도를 증가시키면 Cu2O 상 피크의 세기 는 더욱 줄어들어 결국 농도 X2.5의 전처리용액으로 전 처리 시 Cu2O 상은 거의 관찰되지 않음을 확인할 수 있 었다. 즉, 동일 시간(5분)의 전처리 조건에서 전처리용액 의 농도가 진할수록 전처리 효과는 우수하였으며, X2.5 의 전처리용액 사용 시 Cu2O 상은 관찰되지 않고 순수 한 Cu 상만이 관찰되어 이 농도의 전처리 조건에서 산 화층 제거 전처리가 거의 완전하게 진행되었음을 확인 할 수 있었다. 이러한 결과는 X2.5의 전처리용액으로 앞 서의 산화 동판을 동일하게 전처리한 Fig. 4의 AES 분 석 결과를 통해서도 재확인되었다. 즉, 초기 50 nm에 이 르던 산화층은 X2.5의 전처리용액으로 전처리 시 수 nm 두께 수준으로만 존재함을 확인할 수 있었는데, 이 층 역시도 산소 오염원 및 대기 중 노출에 따라 생성된 자 연발생 산화층으로 판단되는 바 X2.5 전처리용액 사용 시 5분간의 산화층 제거속도는 약 10 nm/min에 근접하 는 것으로 분석되었다. 이와 같이 전처리 후 산화층의 두께가 매우 얇아진 경우에서는 5 nm/min의 AES 에칭 속도 조건으로는 효과적인 분석이 이루어지지 않으므로 0.5 nm/min의 저속 에칭모드에서 분석을 실시해 보았다. Fig. 5는 농도 X2.5의 전처리용액으로 5분간 전처리하여 산화층을 제거한 후 대기 중에서 1일 또는 3일간 노출 시킨 시편을 0.5 nm/min의 에칭속도로 AES 분석한 결 과를 보여준다. 그 결과 각각 약 1 nm 및 2 nm 두께의 산화층이 존재하는 것을 관찰할 수 있었으며, 이를 통 해 Fig. 4에서의 전처리 후 실제 산화층 두께는 1 nm 미 만이고, 그 잔존 산화층은 궁극적으로 자연발생 산화층 일 확률이 매우 높음을 확인할 수 있었다.
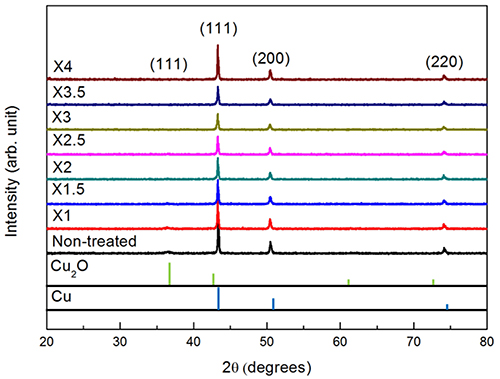
Fig. 3.
XRD patterns in the Cu powders pretreated for 5 min with different concentrations of the pretreatment solution after oxidation in air at 200 °C for 1 h.
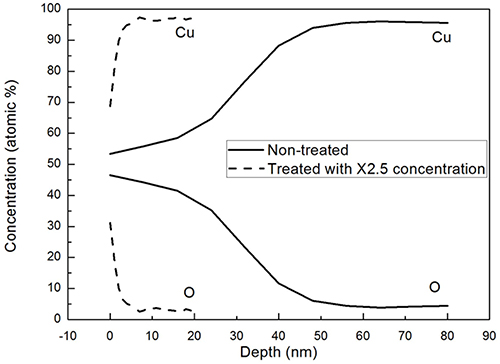
Fig. 4.
AES elemental depth profiles of Cu plates before and after the pretreatment of 5 min with the pretreatment solution of X2.5 concentration after oxidation in air at 200 °C for 1 h.
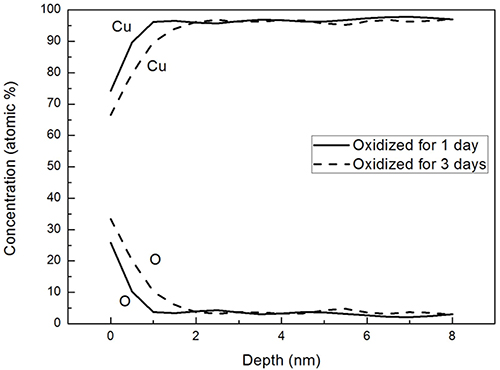
Fig. 5.
AES elemental depth profiles of Cu plates oxidized for 1 or 3 days at room temperature after removal of the oxidation layer.
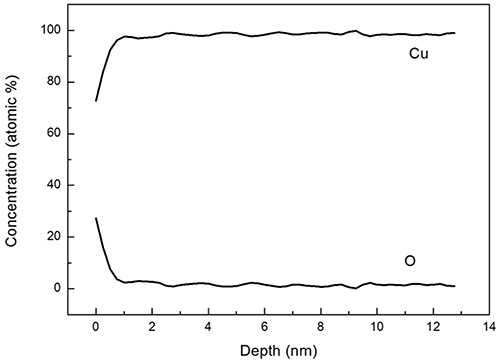
이상의 결과로부터 X1 농도의 전처리용액 사용 시 5 분간의 전처리 동안 약 30 nm의 산화층을 제거할 수 있 고, 그 농도를 2.5배 올린 X2.5 전처리용액을 5분간 사 용할 경우 약 50 nm 두께의 산화층 대부분을 제거할 수 있음을 알 수 있었다. 이러한 결과에 따라 일반적인 Cu 분말의 제조 공정 및 준비 과정에서 생성될 수 있는 약 3 nm 두께 미만 수준의 Cu 산화층을 5 분간의 전처리 로 제거하기 위한 전처리용액의 최적 농도는 언급된 X1 보다 훨씬 묽은 수준이 될 것임을 짐작할 수 있었다. 이 를 검증하기 위해 상온의 대기 중에서 3일간 자연 산 화시킨 동판에 생성된 약 2 nm 두께의 산화층을 X0.13 농도의 전처리용액을 사용하여 5분간의 전처리를 실시한 시편의 AES 측정 결과는 Fig. 6과 같다. 그 결과 앞서 와 마찬가지로 1 nm 미만 두께 수준의 산화층이 검출되 어 농도 X0.13의 묽은 전처리용액은 수 나노미터 수준 의 얇은 Cu 산화층의 제거에 적용 가능함을 확인할 수 있었다.
3.2 동분말의 은도금
본 은도금 공정에서 용해된 silver nitrate로부터 제공 되는 Ag 이온들은 착화제(complexing agent) 또는 킬레 이팅제(chelating agent)로 첨가한 ammonium hydroxide 로부터 제공되는 암모니아와 결합하여 [Ag(NH3)2]+ 형태 의 복합체(complex)를 형성하는 것으로 보고되고 있는 데,13,14) 이 복합체는 이후 전자를 받아 환원되면서 Ag 원자로 전이되게 된다. 이 경우 Ag+ 이온의 산화환원 포 텐샬(redox potential) 값(+0.799 V)은 [Ag(NH3)2]+의 복 합체가 형성됨에 따라 +0.38 V로 감소하여 안정화되므로 이후 반응에서 환원되는 Ag 원자들은 불균일핵생성의 형 태로 생성되는, 즉 Cu 입자 표면을 코팅시키면서 생성 될 확률이 증가하게 된다.15) 200 °C의 대기 중에서 1시 간동안 강제 산화시킨 동분말을 전처리용액을 사용하여 5분간 전처리 시 전처리용액의 농도에 따른 전처리 후 Cu 분말의 SEM 이미지와 Ag 도금을 실시한 Cu 분말 의 후방산란전자(back-scattered electron, BSE) 이미지는 Fig. 7과 같다. X1 및 X4 농도의 전처리용액을 사용한 경우 전처리 직후 Cu 입자들의 이미지는 각각 Fig. 7(a) 및 7(b)와 같았는데, 전처리용액의 농도가 낮았던 Fig. 7(a) 시료의 경우 그 표면이 상대적으로 거친 상태를 나 타내었다. 한편 X1 농도의 전처리용액을 사용한 후 은 도금을 실시한 입자의 이미지는 Fig. 7(c)와 같았는데, 은 도금이 일부 진행되었으나 그 표면이 상대적으로 매우 거칠었고, 미세 입자 형태의 Ag가 Cu 입자 표면에 불 규칙적으로 존재하거나 Cu 입자로부터 분리된 상태로 존 재하여 은도금이 충분히 잘 이루어지지 않았음을 관찰 할 수 있었다. 이러한 결과는 앞서 Fig. 2 결과에서 관 찰되었던 약 20 nm 두께 수준의 잔존 산화층이 Cu 입 자 표면에 존재할 때 환원된 Ag 원자들이 Cu 표면을 균일하게 코팅하지 않고 입자 형태로 균일핵생성된 후 입자 성장 단계에서 Cu 입자 표면에 안착하는 거동에 따른 것으로 분석된다. 단, 각 입자마다 은도금의 정도 나 상태가 크게 차이나는 것으로 관찰되는 바 실제 각 입자에서의 산화층 잔존 정도는 큰 편차가 있을 것으로 판단되었다. 요컨대 Fig. 7(a)에서는 거친 표면은 Cu 산 화층의 불완전한 제거 결과에 기인한 것으로 판단되며, 이러한 잔존 산화층은 궁극적으로 이후의 Ag 도금을 방 해하는 것으로 분석되었다. 반면 X4 농도로 전처리한 후 은도금을 진행한 입자의 BSE 이미지[Fig. 7(d)]를 보면 앞서와 같은 Ag 미세 입자들이 거의 관찰되지 않은 상 태에서 Cu 입자들의 표면이 전이되어 원하는 형태로 은 코팅이 이루어졌음을 예상할 수 있었다. 이후 은코팅의 검 증은 당 분말의 에너지 분산형 X-선 검출 분광기(energy dispersive X-ray spectroscopy, EDS) 분석으로부터 Ag 원소의 검출 결과(Fig. 8)로 보다 명확히 제시될 수 있 었다. 따라서 전처리를 통해 산화층이 거의 제거된 Cu 입자에서는 입자 표면에 은이 전체적으로 균일하게 코 팅될 수 있음을 확인할 수 있었다.

Fig. 7.
SEM images of Cu powders as-pretreated with concentrations of (a) X1 and (b) X4 and BSE images of Ag-coated Cu powders after the pretreatment with concentrations of (c) X1 and (d) X4. Initial Cu powders were compulsorily oxidized in air at 200 °C for 1 h.
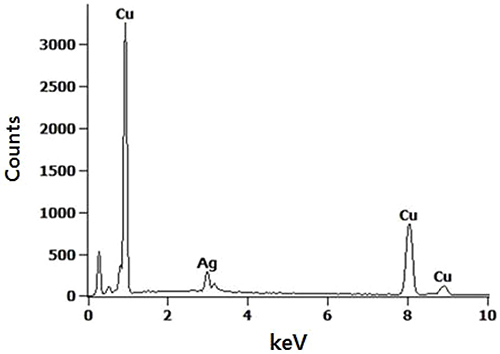
Fig. 9는 상온의 대기 중에서 3일간 노출되어 자연 산 화된 동분말을 은도금할 경우 전처리 여부에 따른 도금 결과이다. 전처리를 하지 않은 상태에서 은도금을 실시한 경우(Fig. 9(a)) Cu 입자의 표면에 얇은 Cu2O 층이 존재 하여 은도금이 거의 이루어지지 않은 BSE 이미지가 관 찰되었고, 일부 Cu 입자 표면에 다양한 크기의 미세 Ag 입자들이 붙어있는 상태가 확인되었다. 이를 통해 약 2 nm의 두께로 예상되는 얇은 산화층이 존재하는 경우 에서도 의도하였던 은코팅은 이루어지지 않음을 검증할 수 있었고, 전처리된 Cu 입자와는 달리 자연 산화로 모 든 Cu 입자에서 얇고 균일하게 산화층이 존재하는 상 태여서 결과적으로 모든 Cu 입자에서 은코팅 결과를 관 찰할 수 없었다. 이와 같은 얇은 산화층을 제거하기 위 해 각각 다른 농도의 전처리용액을 사용한 전처리 공정 후 은도금을 실시한 결과는 다음과 같았다. X0.13 농도 의 전처리용액을 사용한 경우(Fig. 9(b))는 BSE 이미지 상으로 은도금이 된 일부 Cu 입자들을 관찰할 수 있었 고, 미세 Ag 입자들의 크기와 개수가 평균적으로 크게 감소하여 보다 많은 양의 Ag 원자들이 도금층 형성에 소모되었음을 간접적으로 확인할 수 있었다. 즉, 이러한 결과는 수행한 전처리 공정이 산화막 제거에 효과가 있 었음을 보여주나, 모든 Cu 입자에서 산화막의 제거가 완 전히 진행되지 않았음을 나타낸다. 전처리용액의 농도를 X0.3 수준으로 증가시킨 경우의 은도금 결과는 Fig. 9(c) 와 같다. 매우 미세한 Ag 입자들이 코팅된 Cu 입자들 이 일부 관찰되었으나, 궁극적으로 모든 Cu 입자들이 Ag 로 코팅되어 있어 보다 효과적인 전처리 공정으로 더욱 균일한 은도금이 진행되었음을 확인할 수 있었다. 마지 막으로 X0.5의 농도로 전처리를 실시한 경우(Fig. 9(d)) 에서는 미세 Ag 입자들이 거의 관찰되지 않았고, BSE 이미지상으로 모든 Cu 입자들이 Ag로 코팅되었음을 확 인할 수 있어 가장 우수한 품질의 Ag 코팅 Cu 입자가 제조되었음을 관찰할 수 있었다. 이상의 결과들은 Ag 화 학도금액의 장입 직전 Cu 입자 표면에서의 산화층 잔 존 여부가 향후 Ag 도금층의 품질에 매우 큰 영향을 미 친다는 것을 잘 보여준다. 즉, 전처리용액의 농도가 X0.13 으로부터 X0.5로 증가함에 따라 Cu 입자 표면에서의 산 화층 제거가 확실해지고, 이에 따라 환원된 Ag 원자들 은 자체적으로 미세 입자를 형성하기 보다는 불균일핵 생성 과정을 통해 Cu 입자 표면에서의 코팅층 형성에 참여하는 것으로 분석되었다. 단, X0.13 농도의 전처리 용액을 사용한 5분간의 전처리 공정으로 동판 표면에 형 성된 약 2 nm의 산화층을 제거할 수 있었던 Fig. 6 결 과와는 달리 분말을 사용한 Fig. 9에서는 X0.5 농도 수 준의 전처리용액 사용 조건에서 대부분의 Cu 입자 산 화층들을 제거할 수 있음을 관찰할 수 있었는데, 이는 동판에 비해 동분말에서의 산화가 다소 빠른 속도로 진 행될 수 있고, 각 입자마다의 산화 정도도 차이가 나 산 화층이 가장 두꺼운 입자 기준으로 전처리 작업이 요구 되기 때문으로 분석되었다.

4. 결 론
200 °C의 대기 중에서 1시간동안 강제 산화시킨 동분 말과 상온의 대기 중에서 3일간 자연 산화시킨 동분말 을 준비하여 Cu2O 산화층 생성 정도에 따른 전처리 조 건을 제시하였으며, 산화층 제거 정도에 따른 후속 화 학 은도금 공정의 결과를 논의하였다. 강제 산화시킨 동 분말의 경우 X1 농도의 혼합 전처리용액 사용 시 5분 간의 전처리 시간동안 기 형성된 산화층의 두께가 지속 적으로 감소하는 결과를 관찰할 수 있었다. 이러한 결 과는 동일한 조건으로 강제 산화시킨 동판의 AES 분석 을 통해서도 확인할 수 있었는데, 강제 산화로 약 50 nm 에 이르던 산화층 두께는 동일한 전처리 후에 약 20 nm 까지 감소하는 것을 확인할 수 있었다. XRD 분석을 통 해 50 nm 두께의 산화층을 동일한 전처리 시간에 완전 하게 제거하기 위해서는 전처리용액의 농도를 증가시켜 야 함을 알 수 있었고, 이에 따라 5분간의 전처리 공정 을 통해 50 nm 두께의 산화층을 완전히 제거하기 위해 서는 전처리용액의 농도를 X2.5까지 증가시켜야 함을 알 수 있었다. 또한 일반적인 Cu 분말에서 존재 가능한 2 nm 두께 수준의 Cu 산화층은 X0.13 농도의 전처리용 액을 사용한 5분간의 전처리 공정으로 제거 가능함을 확 인할 수 있었다. 그러나 산화층을 제거하는 전처리 직 후 화학 은코팅을 실시하는 실제 제조공정에서 전처리 용액의 최적 농도는 강제 산화 Cu 분말의 경우 X4, 자 연 산화 Cu 분말의 경우 X0.5 수준으로 증가되는 현상 을 관찰할 수 있었는데, 이는 동판에 비해 동분말에서 의 산화가 보다 빠른 속도로 진행될 수 있고, 각 입자 마다의 산화 정도가 차이가 나 산화층이 가장 두꺼운 입 자 기준으로 전처리 작업이 요구되기 때문으로 분석되 었다. 산화층이 충분히 제거되지 않은 전처리 공정 후 은코팅을 실시한 경우 환원된 Ag 원자들이 균일한 코 팅층을 형성하는데 소모되기 보다는 미세 Ag 입자를 형 성하는데 소모되어 원하는 균일한 Ag 도금을 구현할 수 없었다.
