

1. Introduction
2. Basic ML Algorithms
3. Electrocatalysts Discovery with ML
3.1. HER and OER
3.2. HER catalysts discovery with ML
3.3. OER catalysts discovery with ML
4. Opportunities and Prospect
1. Introduction
Fossil fuel consumption and significant greenhouse gas emissions have been endangering the Earth’s ecosystem as a result of rising energy demands around the world.1) To satisfy the rising energy needs and achieve carbon neutrality, there has been a lot of focus on developing sustainable energy technology. The production of renewable energy by electrochemically catalyzed processes, which transform some everyday substances like water, carbon dioxide, and nitrogen into energy carriers, is now a promising technique.2) Among them, hydrogen has received vast attention as a promising alternative energy carrier to replace fossil fuels due to its high energy content compare to that of gasoline. Also, an eco-friendly emission of water vapor instead of greenhouse gas during the combustion of hydrogen is expected to mitigate environmental issues and meet the requirements for circumventing global climate change.
However, more than 90 % of hydrogen is currently produced by reforming fossil fuels, which is called grey hydrogen, and a large amount of carbon dioxide is emitted during the production of grey hydrogen. Despite not producing greenhouse gases, grey hydrogen combustion is not suitable for the overall carbon neutralization. As a sustainable approach, water electrolysis technology has been developed to produce hydrogen without emitting greenhouse gases, which is called green hydrogen.3) A promising method of producing carbon-free hydrogen is electrolysis, which uses electricity to separate water into hydrogen and oxygen. The ideal carbon-free energy ecosystem can be realized if the sources of this electricity are nuclear or renewable resources.
Electrochemical reactions can be divided into two half reactions occurring at the cathode and anode, respectively. When producing green hydrogen through water electrolysis, the hydrogen evolution reaction (HER) and the oxygen evolution reaction (OER) are two separate half processes.4) When enough electrical power is supplied during water electrolysis, the water molecule splits into hydrogen and oxygen gas molecules in a device known as an electrolyzer with two electrodes. Designing excellent electrocatalysts with minimal overpotential is essential to achieving an effective and environmentally friendly hydrogen production in an electrochemical process, which will maximize the conversion efficiency of renewable energy sources.
The development of catalytic materials has relied mostly on empirical, trial-and-error methods due to complicated, multidimensional, and dynamic process nature of catalysis, which eventually requires a significant time and effort to find the most optimized multicomponent catalysts under a variety of reaction conditions. The rational and efficient design of materials with desired performance is the ultimate goal for all researchers in the materials science and engineering field. The most common tactic used by researchers, the classic trial-and-error method, has unavoidably suffered due to the high time and financial costs. The size of the materials space for a search should be constrained due to cost effectiveness, relying on the knowledge of researchers, which has restricted thorough explorations of undiscovered candidate materials.5)
Recently, ML has emerged as a game-changer that could shift a paradigm from a conventional trial-and-error Edisonian approach to data-based predictions.6) ML is commonly used to predict target properties of a material based on the chemical composition, crystal structure of the materials, and basic materials properties [Fig. 1(a)]. For catalyst design, the target properties could be binding energy of adsorbate during intermediate reaction, or conductivity.
The collaborations between machine learning (ML) and catalysts development are rapidly increasing and expanding its area of usage. Historically, the design of materials was based on the intuition and experiment that led to long commercialization timelines. This sluggish traditional progress was the main obstacle to the investment in an early stage of research and also delayed the emergence of the solution for important energy and environment challenges. As computational chemistry has nourished, comprehensive materials databases could be generated in a shorter time frame than the experimental approaches, which becomes the workhorse for the application of ML in materials science. For this reason, computationally calculated materials databases are steadily developed with efficient data structures that help researchers to transform the data into actionable insights [Fig. 1(b)].
To highlight the importance of active electrocatalysts, the fundamental ML algorithms are provided in this review, followed by a brief description of two half reactions, HER and OER. Recent achievements using ML models to discover electrocatalysts for HER and OER by estimating the binding energy of adsorbates for theoretical overpotential prediction are introduced in detail. Challenges on applying ML to catalyst discovery, such as data scarcity and data uncertainty, and current strategies to overcome these challenges are also covered. We believe that our review paper will provide readers with comprehensive and timely information, and that they will benefit greatly from applying ML techniques to their own catalyst discovery.
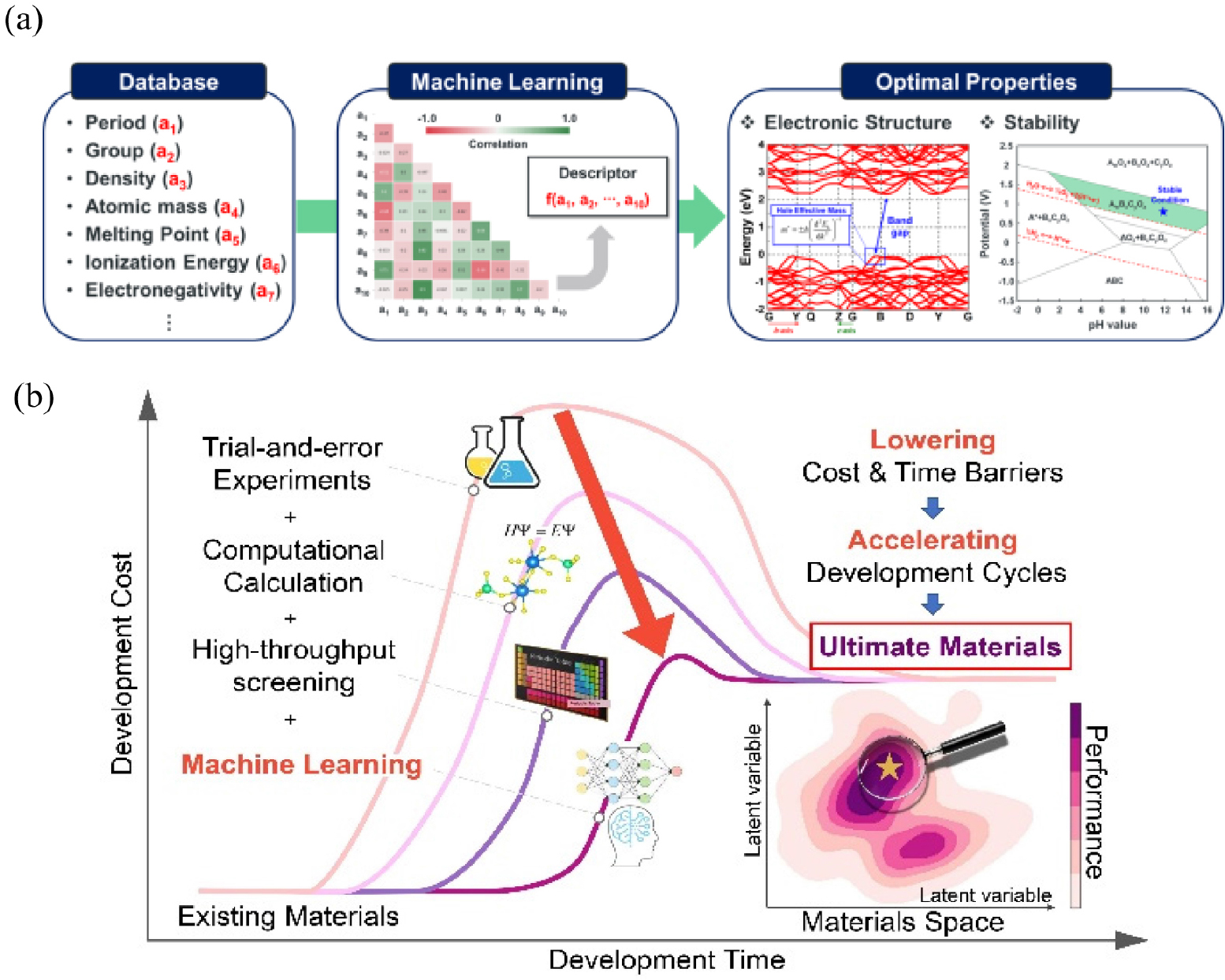
Fig. 1
(a) Schematic of a materials science with ML to predict useful materials science knowledge, such as materials properties, from given database and ML model. (b) Recent trend materials development paradigm in materials science research with ML. Reproduced with permission from Ref. (6). Copyright 2018 American Chemical Society.
2. Basic ML Algorithms
The majority of materials development in the past relied on trial and error. As time went on, scientific theories were methodically established in numerous disciplines, making it possible to produce materials by theoretical calculation. However, the calculations were very difficult and time-consuming. The reactions and properties of materials might be predicted without direct experimentation when more time had elapsed thanks to the invention of computers. The emergence of big data in recent years is a result of the advancement of ICT technology. In light of this, a new paradigm has been proposed, activating data-based research.
It is crucial to extract and process useful information from material data in order to apply big data to actual research and create novel materials.6) ML is one of the most effective techniques. In contrast to conventional learning, ML can create a program from input values and output values (supervised learning). A program can be created utilizing only an input value and no output value if the amount of information used as an output value is minimal (unsupervised learning). Additionally, there is semi-supervised learning, which combines the two teaching approaches. Each learning method has a different learning algorithm, and efficiency can be increased even more by choosing and employing the right algorithm for the job. Supervised learning is fueled by a labeled dataset with known output value. Through input data and labeled data, supervised learning builds a model that can interpret the new data. The amount of training data set must be large to obtain good learning results, and the training data must be generalized.
Support vector machine (SVM) is one of the representative algorithms of supervised learning. It can perform classification or regression by selecting a hyperplane with the maximum margin [Fig. 2(a)]. In particular, it is specialized in classification and is widely used for this purpose. The SVM is based on linear classification and does not consider the dependence between each property. Decision tree (DT) is another method of classifying or predicting the entire data into several subgroups by representing decision rules in a tree-like structure [Fig. 2(b)]. The DT is divided into a classification tree and a regression tree according to which variables are handled. The ensemble of the DTs plays a role as a voting object in random forest that dramatically increase the model accuracy and robustness [Fig. 2(c)].
For successfully extract non-linear input-output relationship, Artificial neural network (ANN) is a learning algorithm created by imitating the learning method of the brain, inspired by the human neuro system [Fig. 2(d)]. Neurons, which are human nerve cells, are composed of dendrites, axons, somas, and synapses. By organizing these neurons into several layers, the weight of the synapse is continuously adjusted so that the training result of the neural network becomes similar to the expected value. This process is called training, and the purpose of training is to find the optimal value of the weight.
Unlike supervised learning, unsupervised learning is not labeled on data. In this case, it may be thought that learning is impossible, but learning is possible. In unsupervised learning, the correct answer is not given, but it is possible to learn by clustering data of similar classes. In order to explain, let us take an example. There are dogs and chickens, and the study can be conducted by dividing dogs with four legs into one group and chickens with two legs into another group. As such, unsupervised learning has excellent utility in classification and has many advantages when performing dimension reduction.
Principal component analysis (PCA) is one of the representative dimension reduction algorithms [Fig. 2(e)]. First, the principal component represented by the linear combination of variables is found by using the variance and covariance relationships between several quantitative variables. A multivariate analysis method that explains most of the total variation with k-th important components is PCA. It usually performed for the purpose of data reduction and interpretation, and it is essential to minimize the loss of information.
Many algorithms are discriminative networks that determine what data is with a certain probability. However, generative adversarial network (GAN) is not discriminant networks but generative models. When learning is performed using training data with a specific probability distribution, data with a similar distribution can be generated. GAN is composed of a generator and discriminator that the learning is progressed by the adversarial relationship between those two agents [Fig. 2(f)].
ML and deep learning is still very active research topic and researchers in worldwide have committed to develop and deploy advanced algorithms with user-friendly interactive tools. Scientists and engineering studying on catalysts should pay attention on cutting-edge ML algorithms like graph neural network (GNN) that has been spotlighted recently to imitate the atomic neighboring environment with node and edges, which is critical to understand the state-of-the-art ML techniques and apply on one’s own research.
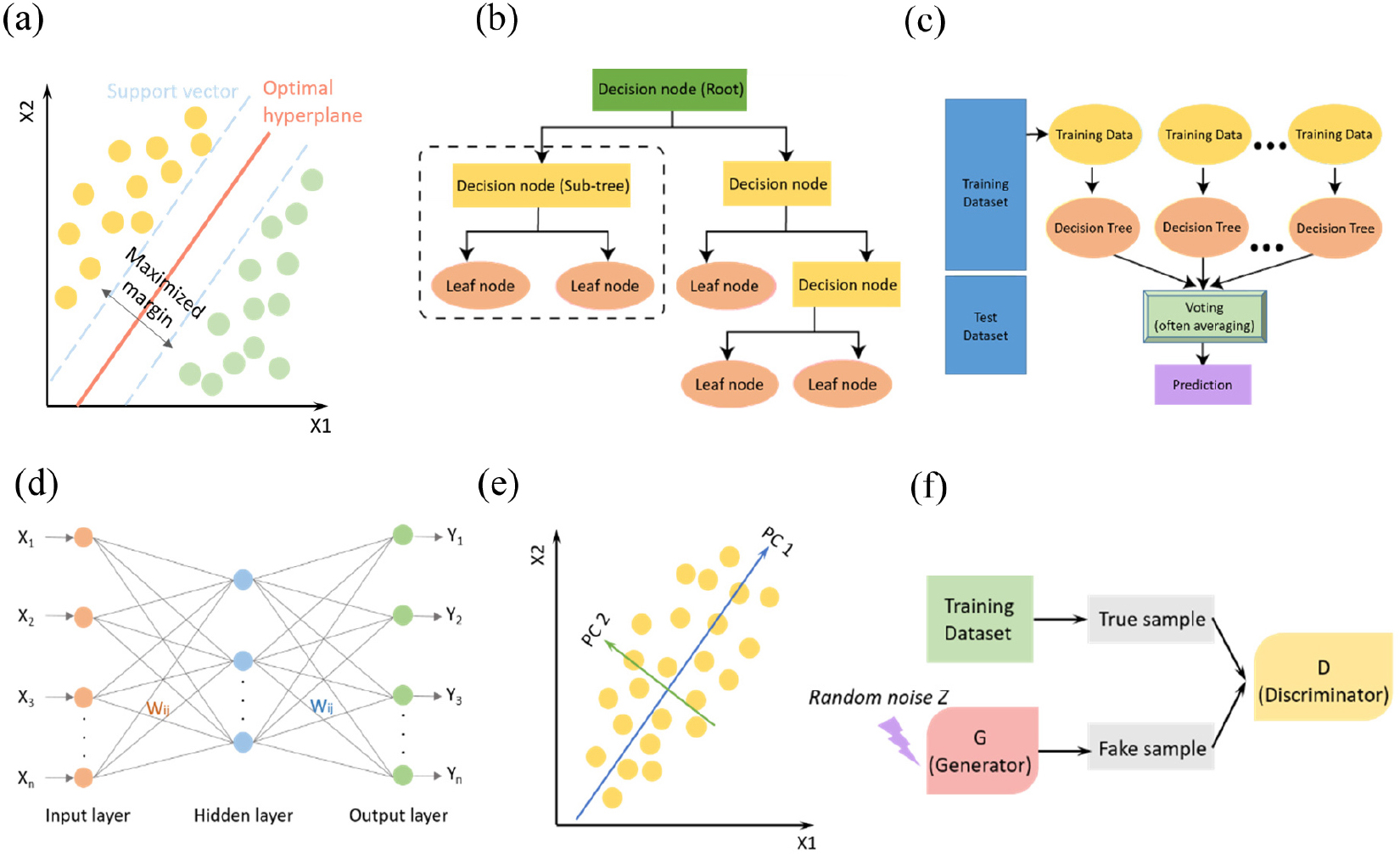
Fig. 2
Graphical representation of supervised (a-d) and unsupervised (e, f) learning algorithms, that is (a) support vector machine (SVM), (b) decision tree (DT), (c) random forest, (d) artificial neural network (ANN) (e) principal component analysis (PCA), and (f) generative adversarial network (GAN) respectively. Adapted with permission from Ref. (6). Copyright 2018 American Chemical Society.
3. Electrocatalysts Discovery with ML
3.1. HER and OER
Electrochemical reactions can be divided into two half reactions occurring at the anode and cathode, respectively.3) In case of water electrolysis for green hydrogen production, HER is a cathodic half reaction and OER is an anodic half reaction. In catalysts working condition, the amount of the required voltage to drive HER and OER is higher than theoretically ideal potential, which are 0 V and 1.23 V vs. RHE, and the difference between onset potential and theoretically ideal potential is defined as the overpotential. To efficiently facilitate water electrolysis and decrease overall cell-working electricity consumption, both HER overpotential and OER potential must be lowered with active catalysts.
The HER is a typical example of a two-electron transfer reaction, and the hydrogen adsorption free energy ΔGH is key for determining activity. Active electrocatalysts for HER should present a ΔGH close to zero, undisturbed adsorption and desorption of adsorbate and H2 product gas. Platinum is the best performing electrocatalyst for the HER because of its thermoneutral ΔGH.
On the other hand, the OER involves a four-electron transfer to oxidize water to oxygen. From pristine active sites on the surface of the catalysts, OH*, O*, and OOH* exist on each intermediate step. The largest energy difference between adjacent steps is the energy barrier for the reaction and become the rate determining step. This energy barrier is related to a theoretical overpotential and should be lowered while having moderate binding energies for the reaction intermediates to be active catalysts. For OER, this trend can be represented with volcano relationship that applies ΔGO -ΔGOH as a descriptor. Both theoretical and experimental overpotential or onset potential can be acquired as target values.
3.2. HER catalysts discovery with ML
In recent years, ML techniques have revolutionized the development and discovery of electrocatalytic and photocatalytic materials in various materials species from metal oxides to single-atom catalysts. The key idea to assess whether highly active catalysts predicting binding energy of adsorbates during the intermediate steps of catalytic reactions. For HER, predict the ΔGH of various materials with pre-trained ML models to screening promising candidate materials with near-zero ΔGH might accelerate the catalyst development process and circumvent tedious trial-and-errors.
Despite the high intrinsic HER activity of Pt, the scarcity and high cost of this precious element limit its commercial use. Researchers have widely developed catalysts to minimize the usage of precious metal or even replace it with the low cost transition metal elements, while maintaining the superior HER activity as Pt. Compared to conventional bulk electrocatalysts, nanomaterials could exhibit even more superior performance as benefited from the rapid development of nanoscience and nanotechnology. Since the binding energy of the reaction intermediate of HER is a key descriptor to theoretically demonstrate the expected HER activity, researchers might efficiently explore a vast chemical space of possible candidates for HER catalysts to replace Pt if machine learning models successfully predict the hydrogen binding energy at the surface of catalysts.
To surpass the superior HER activity of pure Pt metal catalyst, various metal alloys have been investigated to reduce the amount of Pt composition or even exclude Pt. However, the number of possible combinations of elements with different composition is almost infinite, which makes the candidate search difficult. Tran et al. accelerated this candidate searching process by creating a framework combined ML and surrogate-based optimization to guide DFT calculations, which produced 42,785 adsorption-energy calculations to identify 258 candidate surfaces across 102 metal alloys for HER.7) The element type was described by the atomic number, the Pauling electronegativity, the number of atoms coordinated with the adsorbate, and the median adsorption energy between them, and they were further tabulated as the catalyst descriptors for machine learning algorithm. The list of adsorption energy on different surface planes of metal alloy was visualized through t-SNE algorithm that disperse each data point into 2D plane and form clusters from similar neighboring data point. Some Pt-based alloys like PtGa or AlPt, and alloys without Pt like SiV, PdGa, PdSi, or AlNi were predicted to present optimal hydrogen adsorption energy, and those metal alloys could be expected as better candidates for further investigation and experiments.
The metal alloys could be modified by decreasing the size from bulk to nanoparticle, which might increase the surface to volume ratio and enhance a surface catalytic activity. Recently, Mao et al. reported a DFT-based high-throughput screening method combined with machine learning models to successfully identify a type of alloy nanoclusters as the electrocatalyst for HER.8) Cu-based alloy clusters of Cu55-nMn (M = Co, Ni, Ru, and Rh, and n ≤ 22) were optimized and the preference toward core-shell structures with the dopant metal in the core and Cu as the shell atoms was confirmed with Cu-based alloy clusters. The excess energies (E*exc) for Cu55-nMn alloy clusters as a function of the average radial distance of the dopant metal atoms to the core atom were plotted in Fig. 3(a). The lower excess energy indicated a more stable cluster, which led to the optimized atomic ratio of Cu-Ni cluster. With fixed atomic ratio of Cu-Ni cluster, core-shell structure was more stable than segregated cluster structure [Fig. 3(b)]. The neural network model was trained to predict the hydrogen adsorption energy on specific hydrogen adsorption site. The features consisted of the mean bader charge of the first-neighbor and second-neighbor atoms of the active site on the outer atomic shell and coordination number of the active site atom to successfully represent the charge distribution around the active site. The researchers used 3,388 adsorption free energies to predict the H adsorption free energy on nanoclusters with a MAE error of 0.07 eV and a root-mean-square error (RMSE) of 0.11 eV on the test set, which was represented in parity plots [Fig. 3(c)]. The researchers showed that HER performance of the Cu-based nanoclusters can be significantly improved by doping transition metals, which might lead to the optimal shift of hydrogen adsorption energy. Among metal alloy nanocluster candidates, core-shell CuNi alloy clusters were suggested to be the superior electrocatalyst owing to the superior structural stability and the electrochemical activity.
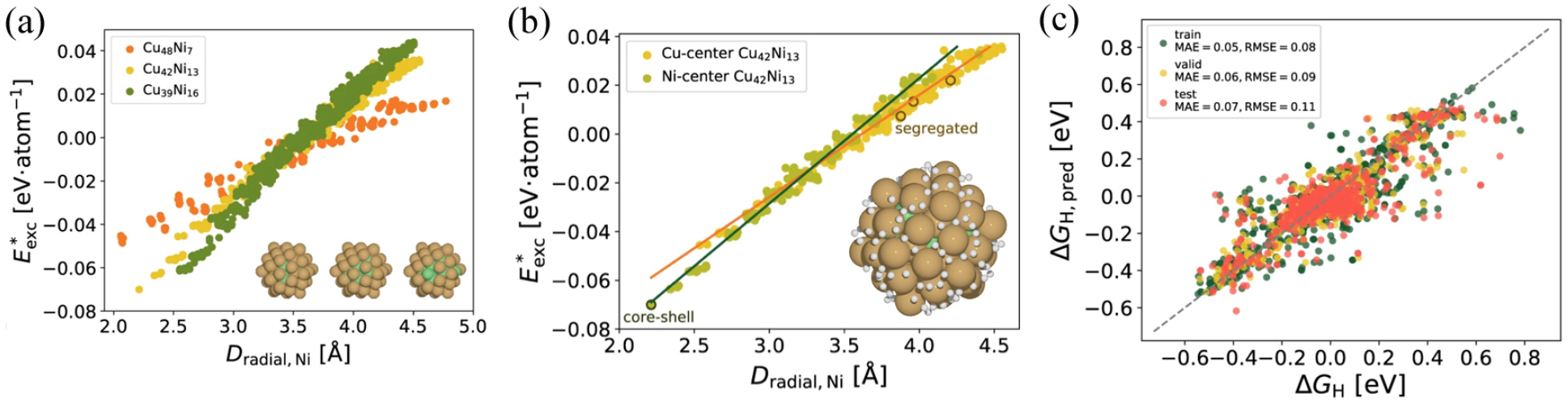
Fig. 3
(a) The excess energies (E*exc) for Cu55-nNin, alloy clusters with n = 7, 13, and 16 as a function of the average radial distance (Dradial, M) between the dopant metal atoms and the center atom. The insets show the lowest-energy configurations. (b) E*exc for Cu42Ni13 alloy clusters as a function of Dradial, M between the dopant metal atoms and the central atom. The core-shell structure and the segregated structure are illustrated. The insets show the optimal adsorption sites for H on the surface of the optimized Cu-centered Cu-Ni core-shell clusters with |ΔGH| < 0.1 eV. (c) Parity plots between predicted and DFT-calculated ΔGH values. Reproduced with permission from Ref. (8). Copyright 2021 Springer Nature.
Similar to nanoparticle, the dimensional reduction of bulk materials into two-dimensional (2D) materials lead to a large surface area to volume ratio the unique physical properties and have been widely researched. This interest led to both a new wave of research on known 2D materials, such as metal dichalcogenides and boron nitride, and the discovery of many new 2D materials. For example, Ge et al. predicted the hydrogen adsorption energy of two single-layer metal dichalcogenides MX2 (M = Mo, W; X = S, Se, Te) in a tilted heterojunction structure by training ML models with rotational angle, bond length, the ratio of the bandgaps of two MX2, and distance between those two layers.9) Using the trained ML model, MoTe2/WTe2 heterojunction with a rotation of 300 degree was predicted to show optimal hydrogen adsorption behavior towards efficient HER and suggested as a promising HER catalysts.
Another widely investigated 2D materials species is 2D MXenes, which is a 2D flake of carbides and nitrides of transition metals. The general formula of MXenes is expressed as Mn+1XnTx (n = 1, 2, 3), where M is an early transition metal, X is C or/and N, and Tx is the surface functional group, such as -O, -OH, and -F. 2D MXene materials have attracted the researchers due to their advantages of adjustable chemical composition, tunable layer thickness, and facile functionalization nature with outstanding physiochemical properties, which could be beneficial for HER catalysts design. However, tuning the thermal stability and activation of in-plane activity still remain as a challenge. To solve this problem, Zheng et al. built a machine learning framework combined with screening based on DFT calculations to predict hydrogen adsorption energy on MXenes with various hydrogen coverages.10) Various bare MXenes without hydrogen coverage were modelled to obtain the hydrogen adsorption free energy values by a series of DFT calculations [Fig. 4(a)]. Different numerical intervals of ΔGH* were represented with the different colored circles.
To tune the hydrogen adsorption, the S functional group was introduced onto the bare MXenes. 20 primary descriptors were suggested to efficiently represent the hydrogen adsorption properties on MXenes and the correlation map of those features was used to narrow down the number of descriptors [Fig. 4(b)]. Features with top 10 importance were selected and it turned out that the electronegativity and atomic mass of transition metal element were important to increase the accuracy of machine learning model performance on predicting the hydrogen adsorption energy of the bare MXenes [Fig. 4(c)]. Also, the charge ratio of transition metal elements and of atomic radius corresponded the electronic and geometric structures that could be applied as key descriptors for the machine learning model to predict the hydrogen adsorption energy on the MXenes with a wide range of hydrogen coverages. The random forest regression model showed the lowest test set error compared to support vector or kernel ridge regression model and the best perfoming random forest regression model was selected to predicted that under wide hydrogen coverages. Os2B- and S-terminated Scn+1Nn (n = 1, 2, 3) MXenes exhibited optimal hydrogen adsorption free energy near zero, which leads to a superior HER activity with the ΔGH value approaching zero under wide hydrogen coverages [Fig. 4(d)]. DFT calculations on different hydrogen coverages on the promising candidates were conducted to verify the prediction from the machine learning model, and the author claimed that S functional groups play a crucial role in regulating the HER performance due to the antibonding states which are full of electrons.
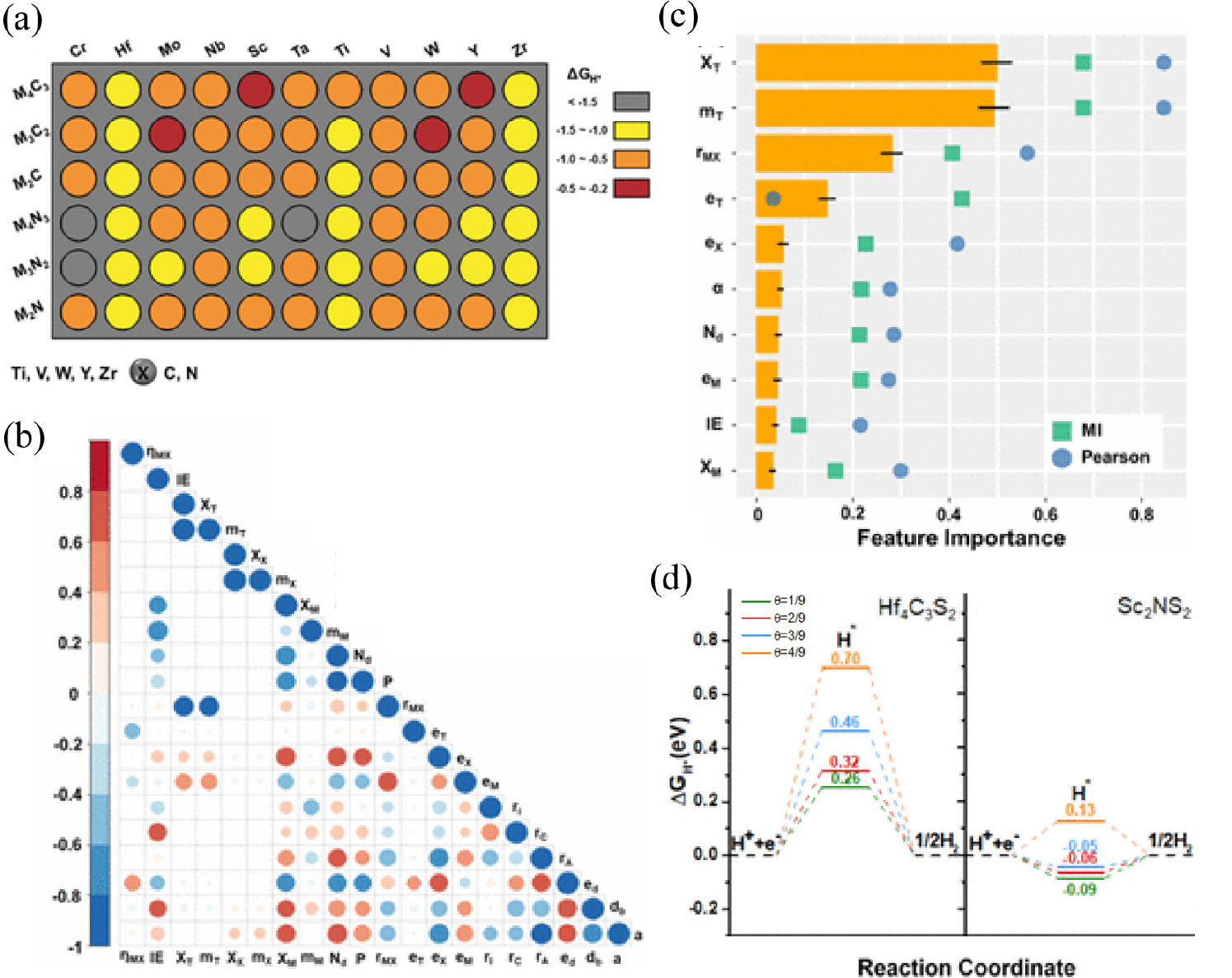
Fig. 4
(a) Color block map of ΔGH* for the bare MXenes. Gray, yellow, orange, and wine-red circles represent ΔGH* values of < -1.5, -1.5~-1.0, -1.0~-0.5, and -0.5~0.2 eV, respectively. (b) Correlation map of 20 features, which resulted in red for the strong positive correlation, blue colors correspond to the strong negative correlation, and white means no direct correlation between two descriptors. (c) Importance of top descriptive 10 features suggested from the random forest machine learning models. (d) ΔGH* of Hf4C3S2 and Sc2NS2 with different adsorbed hydrogen coverages from 1/9 to 4/9. Reproduced with permission from Ref. (10). Copyright 2020 American Chemical Society.
Furthermore, Wang et al. expanded the chemical space of O-terminated MXenes to the ordered binary alloy based MXenes with different combination of two 3d, 4d and 5d transition metal elements, which leads to M2M′X2O2 and M2M′2X3O2 systems (X = C or N).11) The 110 kinds of experimentally unexplored 2D MXene OBAs from 2,520 candidates were predicted to indicate with outstanding thermostability and HER activity. The geometrical and electrical descriptors were selected to link the improved HER activity with the alloying effect through the ensemble learning model. The hydrogen adsorption energy on ordered binary alloy based MXenes covered with hydrogen was predicted with the machine learning model and Ti2M′2C3O2 (M′ for 3d = Ti, V, Cr; 4d = Zr, Nb, Mo; 5d = Hf, Ta) were expected to show exceptional HER activity due to their optimal hydrogen adsorption strength. Sun et al. also expanded the candidate group to MBenes that could be represented as MmBnTx (m, n = 1, 2), and trained machine learning models to successfully predicted the hydrogen adsorption energy. As a result, Co2B2 and Mn/Co2B2 were highlighted as the excellent HER catalysts due to |ΔGH* | < 0.15 eV over a wide range of hydrogen coverages.
2D materials could be applied as an efficient catalyst on not only electrocatalytic water splitting but also photocatalytic water splitting. Photocatalytic water splitting is also promising alternative for the generation of hydrogen without using fossil fuel. Photocatalysts based on bulk oxide materials such as TiO2 suffered from large band gaps, low harvesting of visible light, and high tendency for charge recombination.12) To circumvent those problems, various classes of 2D materials have been developed to offer several advantages such as increased active sites per surface area, enhanced charge separation and transport over their bulk counterparts. Kumar et al. devised interpretable machine learning model to find efficient 2D water-splitting photocatalysts, based on 3,099 2D materials belong to the octahedral symmetry group (Oh) or 1T phase, in which six hydroxyl ligands are attached to a metal atom in the octahedral geometry.13) The total computational time required to perform the relevant DFT calculations amounted to about three years or 450,000 CPU core hours, justifying the need for an ML-based study to quickly screen new stable 2D materials. Hence, highly accurate ML methods, including mean feature ranking and Bayesian hyperparameter optimization were performed to predict formation energies and convex hull distances. The best three hyperparameters for the formation energy regression were represented in contour plots, consisted with learning rate, number of leaves in random forest models, and lambda factors [Fig. 5(a)]. After ML prediction for phase stability, a funnel-like screening procedure in Fig. 5(b) was conducted. The first tier screened 3,099 octahedral 2D (2DO) materials in database based on their overall stability descriptors. The second tier selected semiconductors with specific symmetrical structure and having PBE band gaps between 0.5 and 2.0 eV. The selected 37 2DO materials were finally screened based on suitable GW band gaps and band alignment.
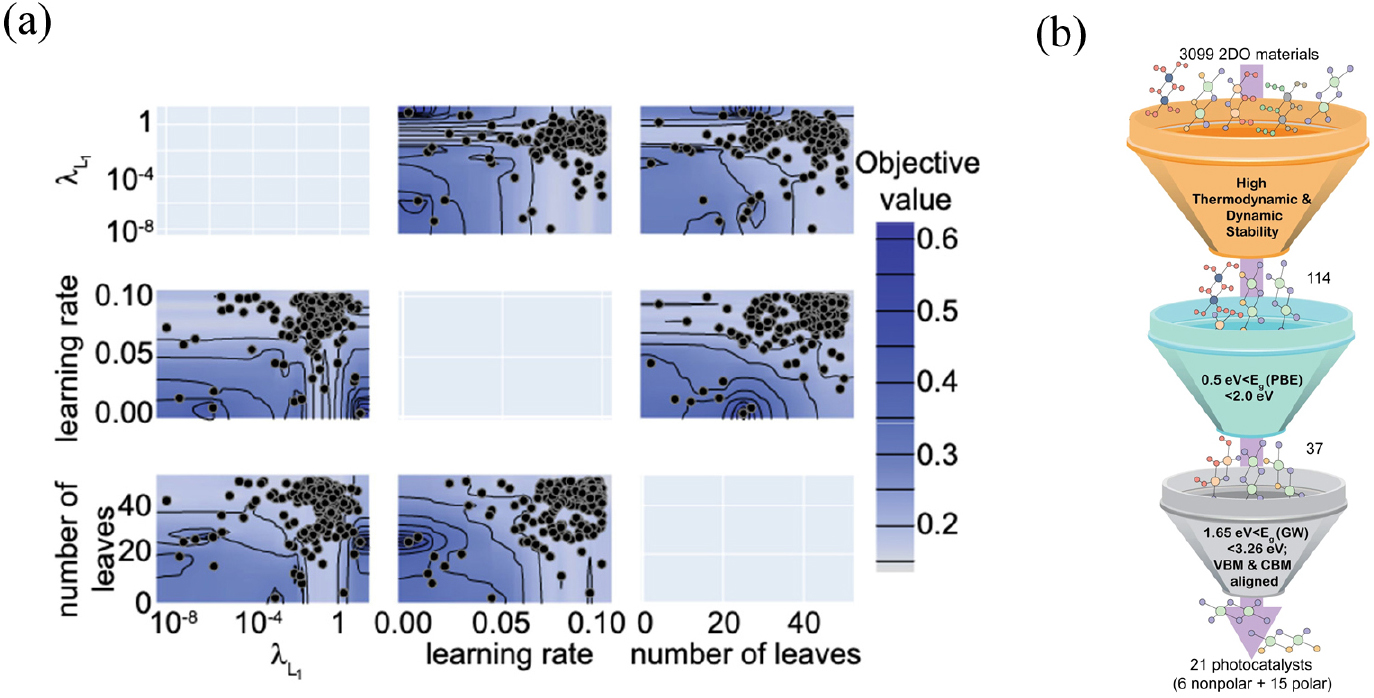
Fig. 5
(a) Contour plot of best three hyperparameters utilized in the LightGBM algorithm-based ML model using selected features for the formation energy regression (b) The high-throughput scheme utilized for screening stable and catalytically active 2DO photocatalysts with (thermo) dynamic stability, suitable PBE band gaps, and GW bandgap with band alignments. Reproduced with permission from Ref. (13). Copyright 2021 Springer Nature.
The most stable 2D materials were further screened based on suitable band gaps within the visible region and band alignments with respect to standard redox potentials using the GW method, resulting in 21 potential candidates. HfSe2 and ZrSe2 were found to have high solar-to-hydrogen efficiencies reaching their theoretical limits. These suggested 2D materials are also expected to mitigate the viability of charge carrier recombination by reducing the distance required for photogenerated electrons and holes for reaching the active sites. Researchers also confirmed with further calculation on target material system to evaluate the band gap position and connect with the localization of CBM and VBM to the optimal photocatalytic advantages.
Noble nanoparticle (NP)-sized electrocatalysts have been exploited for diverse electrochemical reactions including HER to facilitate the water electrolysis to realize the eco-friendly hydrogen economy. For further improvement in activity and cost effectiveness, minimal amounts of single atoms have been recently exploited to maximize the active surface area and to tune the catalytic activity by coordinating the single atoms in defect sites of N-doped graphene.14) To explore the optimal single-atom electrocatalysts, Ha et al. screened single atom catalysts with 3d-5d transition metal elements using DFT along with ML-based descriptors.15) The stability and activity of M-N-doped graphene were explored from the view of structure/coordination, formation energy, structural/electrochemical stability, electronic properties, electrical conductivity, and reaction mechanism. Among various –NnCm moieties, the –N2C2 moieties show higher electrochemical catalytic performance and longer durability (without aggregation/dissolution) compared with the widely studied pure –C4/C3 and –N4/N3 moieties, and also the formati–n of -N2C2 moieties were also energetically favored than other moieties [Fig. 6(a)]. The ML-based descriptors were carefully selected to predict hydrogen adsorption energy with low error to discover better catalysts than benchmark noble metal catalysts. In the N2C2 templates, Ni/Ru/Rh/Pt single atom active sites were predicted to show low HER overpotentials due to their optimal hydrogen binding energy near zero.
The catalytic activity was investigated in terms of H-adsorption binding/free energies for the intermediates during the HER. HER overpotential (ηHER) was calculated based on the ΔGH*, which is used to find outstanding HER catalysts, and visualized with representative colormap [Fig. 6(b)]. Mo/Rh/La/Os/Pt in C4-graphene, Ti/Zr/Ag/Cd in N1C3-N-doped graphene, Co/Ni/Ag in N2C2b-N-doped graphene, Cr/Cu/Cd in N3C1-N-doped graphene, Rh/Cd in N4-N-doped graphene, and Ni/Ru/Rh/Pt in –N2C2-N-doped graphene templates show -ηHER < 0.1 V and are likely to be super-performing HER catalysts, given that the Pt(111) surface shows -ηHER = 0.2 V on the fcc-hollow site. Indeed, Pt-N2C2, Ru-N2C2, and Rh-N2C2 were experimentally synthesized and reported as remarkably active HER catalysts. The advanced research strategy, which combines high-throughput computing with machine learning, shows a dramatic acceleration of catalysts search and ensure robust ability for evaluating unexplored new types of materials as a targeted electrocatalysts.
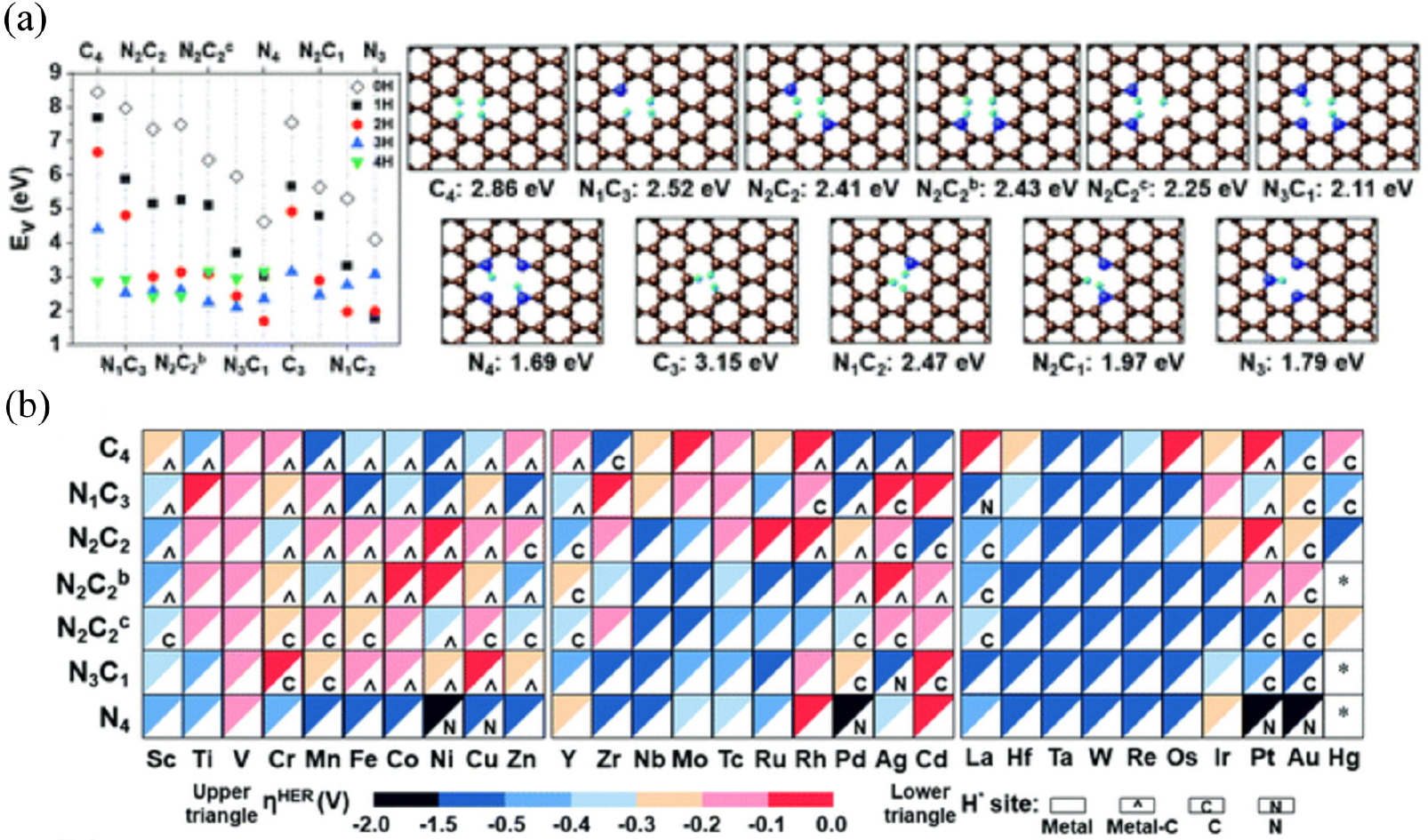
Fig. 6
(a) DFT-calculated vacancy forming energies for –NnCmHh moieties of N-doped graphene in the presence of H but in the absence of metal atoms. (b) HER overpotentials (ηHER) of Metal-NnCm on N-doped graphene. Color code map for ηHER (upper triangle) and most stable H-adsorption sites (H*) (lower triangle), which are denoted by metal site (in white blank), metal-C bridge site (by a hat symbol ‘‘^’’), C site (by ‘‘C’’) or N site (by ‘‘N”). Reproduced with permission from Ref. (15). Copyright 2021 Royal Society of Chemistry.
3.3. OER catalysts discovery with ML
Compared to the two electrons transfer process in HER, OER has four intermediate steps with total four electrons transfer process, which leads to unavoidable sluggish kinetics that greatly reducing the energy efficiency of the overall reaction. Significant efforts have been made over past decades to develop highly active electrocatalysts to improve the reaction rate of OER. Currently, the state-of-the-art OER catalysts are metal oxides based on the precious element the metal, such as RuO2 and IrO2. For OER, predict the ΔGO and ΔGOH on the catalyst surface with ML model to locate the top of the volcano plot leads to promising candidate materials with lowest overpotential. To accelerate the catalyst screening and rationalize the catalyst design, revealing the intrinsic factors that affect the OER activity of multicomponent transition metal oxides is the key.
To fully commercialize the highly efficient proton exchange membrane (PEM) electrolyzers produce hydrogen with higher purity, superior electrocatalyst with economical, active, and acid-stable catalysts for the OER occurring at the anode. Discovering acid-stable, cost-effective, and active catalysts for oxygen evolution reaction is critical since this reaction is a bottleneck in many electrochemical energy conversion systems, including electrochemical water electrolysis. The current systems use extremely expensive iridium oxide catalysts. Identifying Ir-free or less-Ir containing catalysts has been suggested as the goal, but no systematic strategy to discover such catalysts has been reported. Back et al. performed first-principles-based high-throughput catalyst screening to discover OER-active and acid-stable catalysts focusing on equimolar bimetallic oxides with space groups derived from those of IrOx.16) The researchers developed an approach to evaluate acid-stability under the reaction condition by utilizing the Materials Project database17) and DFT calculations. Acid-stable materials were further investigated with their OER catalytic activities and identify promising OER catalysts that satisfy all the desired properties: Co-Ir, Fe-Ir, and Mo-Ir bimetallic oxides.
Moreover, machine-learning-based surrogate models have the potential to accelerate the search for polymorphs that target specific applications. Recently, a generalizable active learning accelerated algorithm for identification of electrochemically stable iridium oxide polymorphs of IrO2 and IrO3 (Fig. 7).18) The search was combined with a subsequent evaluation of the structures’ electrochemical stability for the acidic oxygen evolution reaction. By finding all 956 structurally distinct AB2 and AB3 prototypes in current materials databases, more than 38,000 of structural candidates were generated. The researchers confirmed the overall stability of the rutile structure by discovering 196 IrO2 polymorphs within the thermodynamic amorphous synthesizability limit and employing an active learning methodology. A random search of the candidate space was done to test the algorithms performance and at least a 2-fold increase in the rate of discovery was confirmed. Additionally, the active learning approach can acquire the most stable polymorphs of IrO2 and IrO3 with fewer than 30 density functional theory optimizations [Fig. 8(a)]. Analysis of the revealed polymorphs’ structural characteristics suggests that almost all low-energy structures prefer octahedral local coordination settings [Fig. 8(b)]. A subsequent Pourbaix diagram on the Ir-H2O system revealed that rutile IrO2 was no longer as stable as α-IrO3 under acidic OER conditions [Fig. 8(c)]. Also, the DFT calculation of theoretical OER surface activities demonstrated an ideal weaker binding of the OER intermediates on α-IrO3 than on any other considered iridium oxide. The proposed active learning algorithm could be easily generalized to search for any binary metal oxide structure with a defined stoichiometry.
To expand the catalysts candidate species to the bimetallic perovskite structure, Li et al. develop an adaptive machine learning strategy in search of high-performance double perovskites (AA′B2O6) modified ABO3-type cubic perovskites for catalyzing OER [Fig. 8(a)].19) An exploratory analysis by comparing the overpotential distribution of the candidates with different B-site elements was conducted to gain compositional insights into the OER activity [Fig. 8(b)]. A set of multifidelity features such as composition and electronic structure as optimized and probabilistic models with Gaussian processes were trained with DFT calculated *O and *OH adsorption energies as catalytic activity descriptors. By setting criteria of the theoretical overpotentials to be less than 0.5 V, candidates were iteratively refined and investigated throughout ML models with small RMSE less than 0.5 eV. This ML model could rapidly navigate through a chemical subspace of ~4,000 AA′B2O6 and single out stable structures with promising OER activity [Fig. 8(c)]. Various known perovskites with improved catalytic performance over the benchmark LaCoO3 were successfully identified [Fig. 8(d)] and also other additionally unexplored candidates were suggested for further investigation.
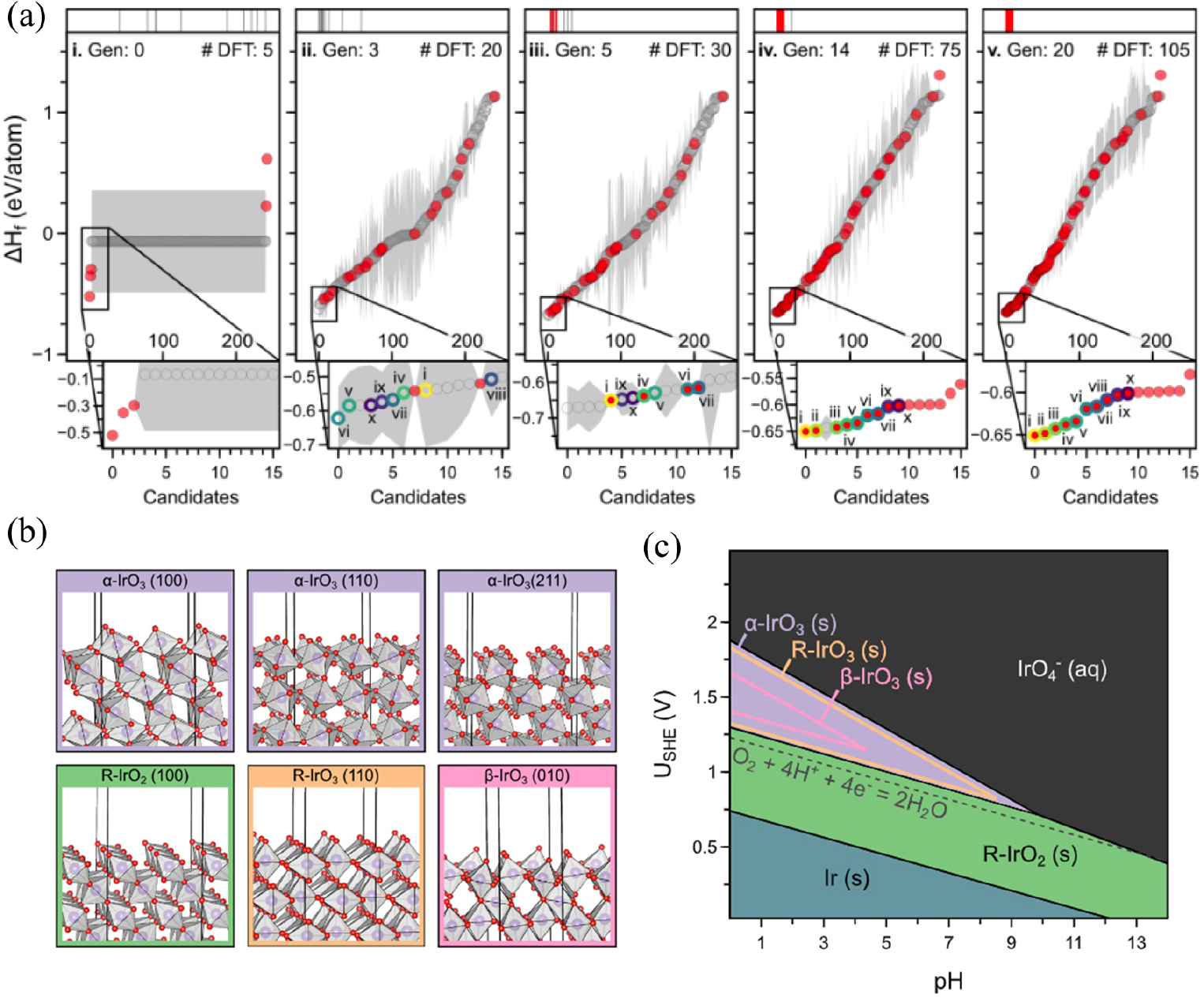
Fig. 7
(a) For five different generations on active learning process for searching the electrochemically stable iridium oxide polymorphs. (b) Structural models with one monolayer of adsorbed O atoms on the different surface of IrOx polymorphs. (c) Pourbaix diagram of Ir-H2O system demonstrated as a function of applied potential (USHE) and pH of the electrolyte. Reproduced with permission from Ref. (18). Copyright 2020 American Chemical Society.
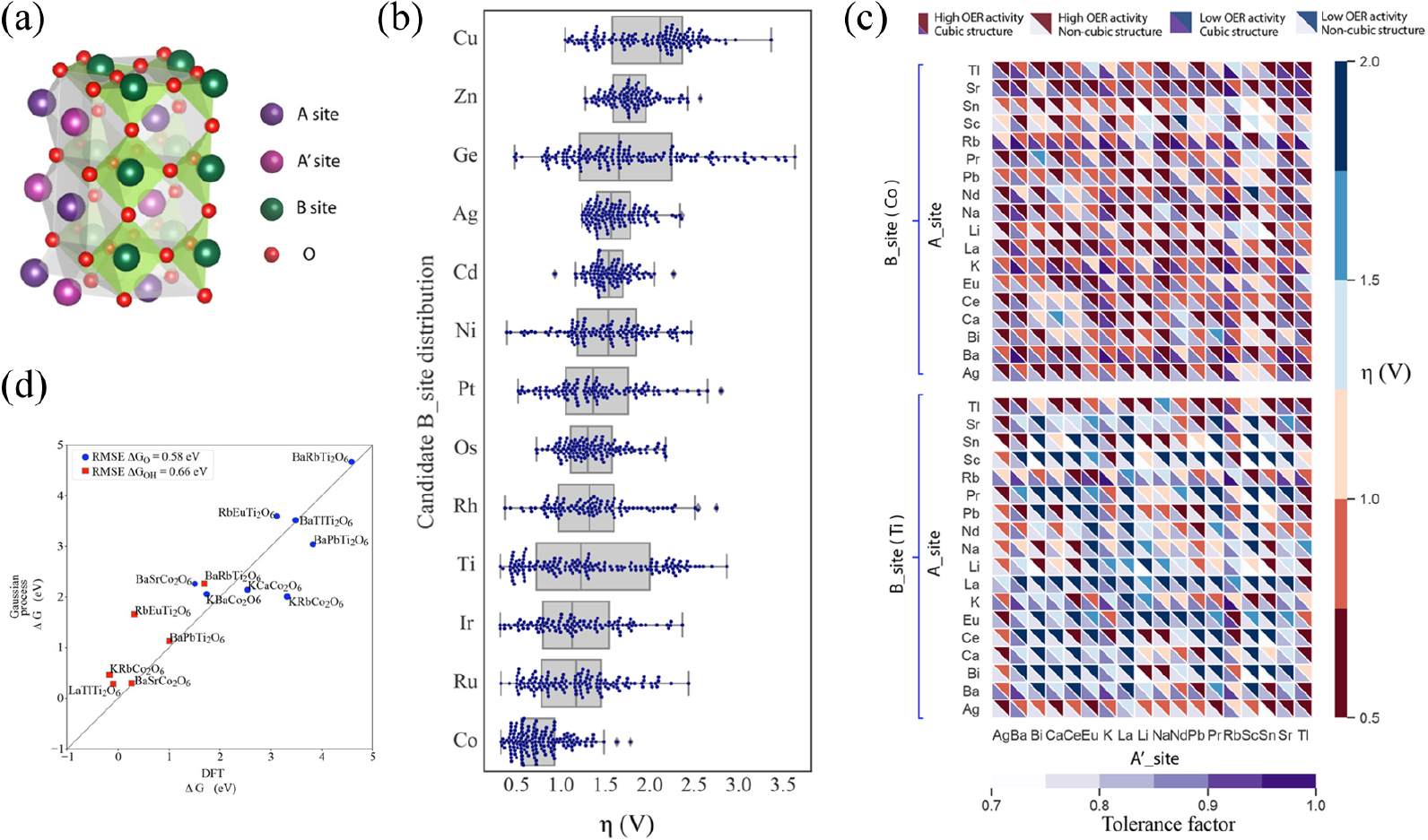
Fig. 8
(a) Configuration of AA′B2O6 cubic double perovskite. (b) Box and swarm plots display of the overpotential distributions for the candidate data set with different B-site metals. (c) Heat map visualization of the OER activity of double perovskites as a function of A-site/B-site cations in terms of the OER overpotentials and cubic phase probability. The red/blue color bar denotes the overpotentials, and the purple bar represents the tolerance factor. (d) Parity plot for DFT-calculated versus Gaussian process model prediction of descriptor adsorption free energies on candidate perovskite structures. Reproduced with permission from Ref. (19). Copyright 2020 American Chemical Society.
For another mathematically reasonable approach on descriptor development, symbolic regression (SR) is a promising method of interpretable machine learning for building mathematical formulas that best fit certain datasets. Recently, Weng et al. used SR to guide the design of new oxide perovskite catalysts by developing a simple descriptor, μ/t, where μ and t are the octahedral and tolerance factors, respectively.20) With a simple descriptor, the discovery of a series of new oxide perovskite catalysts with various elemental ratio was accelerated that enabled the expansion of candidate chemical spaces. The researchers then synthesized five new oxide perovskites and characterized their OER activities. Four of them, Cs0.4La0.6Mn0.25Co0.75O3, Cs0.3La0.7NiO3, SrNi0.75Co0.25O3, and Sr0.25Ba0.75NiO3, were among the oxide perovskite catalysts with the highest intrinsic activities, which highlighted the predictive power of SR on OER catalysts screening process.
To maximize the atomic utilization, atomically dispersed single atom catalysts recently gain attentions from researchers. Recently, Wu et al. designed a topological information-based ML model to map the OER overpotentials with atomic properties of the corresponding SACs.21) A topology-based ML-accelerated prediction of OER overpotential of all transition metals was reported based on DFT calculations of 15 species of SACs. The trained ML model enabled a 130,000-fold reduction of prediction time compared to traditional DFT calculation. It also yielded remarkable prediction precision with a low relative error of 6.49 %, which assured both prediction accuracy and time efficiency at the same time.
For solar-driven photoelectrochemical water splitting, doping is an effective strategy for tuning metal oxide-based semiconductors, since the proper bandgap leads to an optimal light absorbance. Despite years of intensive research, choosing the right dopant is still primarily based on trial and error. Because it can identify correlations from the ostensibly unclear relationships between a wide range of dopant characteristics and the PEC performance of doped photoelectrodes, machine learning is promise in delivering predicted insights on the dopant selection for high-performing PEC systems. Recently, Wang et al. successfully built ML model to predict the doping effect of 17 metal dopants into hematite structure of Fe2O3, a prototype photoelectrode material.22) The critical parameters from the 10 intrinsic features of each dopant were extracted and validated experimentally by the coherent prediction on Y and La dopants’ behaviors. From the ML model, the chemical state was selected as the most significant selection criteria and dopants with higher metal-oxygen bond formation enthalpy and larger ionic radius were favored in improving the charge separation and transfer in the Fe2O3 photoanodes. The generic feature of this ML guided selection criteria was further extended to CuO-based photoelectrodes by alkaline metal ions doping. Those ML-assited dopant searching process could be easily transferred to other types of catalysts design, such as transition-metal based layered double hydroxides.23)
4. Opportunities and Prospect
ML is undoubtedly revolutionizing the whole paradigm in almost every fields, including science and engineering. Those recent surge of interest in ML could mislead to the wrong statement that ML is an absolute solution for every single case. In fact, however, ML is not needed in many cases, and even show worse performance on some tasks. Researchers should especially remind themselves with a standard whether should or should not use ML in given task. ML might be a meaningful strategy for an appropriate problem that human often struggles with, such as when the data and their interactions too complex and could not be interpreted.
There are several challenges in applying ML to materials science and engineering. The most frequent difficulty for researchers who want to apply ML to the materials field is the small amount of data. Since the absolute amount of data related to ongoing research is tiny, it is often difficult to proceed with ML based on this data. However, some state-of-the-art machine learning techniques, such as few-shot learning or transfer learning, have been extensively applied to overcome the data deficiency and expand the domain of ML-assisted advances in materials science and engineering fields.
When the training data and search space covered distant crystal structures and chemical compositions, it was challenging to directly use the elemental and structural representations as descriptors. In particular for energic indicators, such as the adsorption energy of O or H species on HER and OER electrocatalysts and the binding energies of electrocatalysts with the supports, the results of DFT calculations have been stored as databases for prediction and classification of target physical properties in ML thanks to advanced techniques and computational power.
There are now a few extensive databases that provide DFT calculation data for computational catalysis research. For instance, Catalysis-Hub is a web-platform allowing comprehensive and user-friendly access to heterogeneous catalysis concepts, whose surface reactions database contains thousands of reaction energies and barriers from DFT calculations on surface systems.24) Open Catalyst Project is another collaborative research effort for using AI to model and discovering new catalysts that contain 1.2 million molecular relaxations with results from over 250 million DFT calculations.25) With the help of ML and big data, discovery towards complex multicomponent catalysts for the optimal reactions might be accelerated. For example, transition metal-based alloy,26) atomically dispersed dual-atom catalysts27) or 2D materials,28) The ML-assisted catalysts development paradigm might be expanded to various electrochemical reactions, such as water oxidation under neutral condition,29) CO2 reduction,30) ammonia oxidation,31) and photoelectrochemical reactions.32) We believe that ML will unlock the limitation on chemical space for catalysts search and superior catalysts for efficient water electrolysis could be discovered and synthesized,33) which might accelerate the green hydrogen ecosystem with a large-scale hydrogen production.34)
