

1 Introduction
Metal oxides are commonly used in industrial applications as catalysts or as support materials. Many properties of metal oxide surfaces have been studied during the past decade.1-3) CaO is a metal oxide, commonly known as quicklime or burnt lime, is a widely used chemical compound. It is a white, caustic, alkaline, crystalline solid at room temperature investigated by Broqvist, et al.4) Moreover; CaO can be an effective destructive adsorbent for small compounds such as fluoro-, chloro-, and bromocarbons, sulfur- and organophosophorus compounds. 5) Coal-fired power plants are the greatest anthropogenic source of mercury emissions.3) In this work, Density Functional Theory(DFT) with cluster methods were predominantly used to simulate the CaO (001) surface. The DFT scheme has been proven to be fairly accurate.6-10) The number of published papers employing CaO cluster models is quite limited, but some publications have appeared.11,12) Recent work on a similar cluster looked at NOx and other compounds being adsorbed on a relatively small CaO cluster.13) Several researchers used a fixed cluster approach.11-14) There are some calculations on CaO surfaces, which are for much smaller adsorbents where more elaborate methods, can be applied.11-17) Nanomaterials are of great scientific interest due to their size-related unique properties and a wide variety of potential applications. However, a common phenomenon of metal oxides during preparation is uncontrolled agglomeration and growth of large particles. The aggregation nanomaterial results in the growth of nanoparticles often to sizes in excess of the nanoparticle size definition of less than a 100 nm, ultimately leading to sedimentation. The aggregated particles differ significantly from nanomaterials for which size-dependent properties are observed. This growth can be prevented by the presence of surfactants and other surface coatings. Therefore, synthesis of metal oxide nanostructures with controlled size, uniform morphology, good crystallinity and high dispersion remains a major challenge.
Mercury is a toxic metal that accumulates in the food chain and is considered a hazardous air pollutant. Mercury has severe health effects and may cause central nervous system damage, pulmonary and renal failure, severe respiratory damage, blindness and chromosome damage. The Hg emissions from coal-fired power plants are of major environmental concern. Mercury in coal is vaporized into its gaseous elemental form throughout the coal combustion process. Elemental Hg can be oxidized in subsequent reactions with other gaseous components and solid materials in coal-fired flue gases. While oxidized Hg in coal-fired flue gases is readily controlled by its adsorption onto fly ash and/or its dissolution into existing solution-based sulfur dioxide scrubbers, elemental Hg is not controlled. The extent of elemental Hg formed during coal combustion is difficult to predict since it is dependent on the type of coal burned, combustion conditions, and existing control technologies installed. Therefore, it is important to understand heterogeneous Hg reaction mechanisms to predict the speciation of Hg emissions from coal-fired power plants to design and effectively determine the best applicable control technologies; specifically Mercuric chloride. HgCl2 is highly toxic, both acutely and as a cumulative poison. CaO can act as adsorbent of Hg-containing species which will appear in subsequent sections. Specifically, HgCl2 adsorption on the CaO (001) surface was studied by performing DFT calculations using B3LYP functionals. However, it should be pointed out that the accuracy of DFT in treating the Hg transition metal system does have limitations. The Hg system possesses spin multiplet structures that are energetically close to each other.9,10)
The most prominent greenhouse gas is carbon dioxide. Coal-fired power plants and fossil fuel combustion is also the major source of carbon dioxide emissions, there is difficulty in controlling carbon dioxide in flue gases because of the large quantities of carbon-content in fossil fuels. In order to derive novel adsorbents for removing carbon dioxide, it is important to understand the adsorption mechanisms of CO2. Graphene is a thin layer of pure carbon; it is a single, tightly packed layer of carbon atoms that are bonded together in a hexagonal honeycomb lattice. Recent advances suggest that reduced graphene serves as a good 2-Dimensional (2D) support to nucleate metal oxide nanoparticles on the edges and surface.18) It has been demonstrated that graphene can serve as a perfect 2D support for anchoring metal or metal oxide nanoparticles.18)
The mechanism of mercury and CO2 adsorption on CaO is not exactly known and its understanding is crucial to the design and fabrication of effective capture technologies for mercury and CO2. In this work, theoretical calculations have been performed to investigate the adsorption of Hg and CO2 on the combined advantages of both graphene and metal oxides solid surfaces(g-CaO) for improving the HgCl2 capture. The objective of this research is to identify potential materials as multipollutant sorbents in power plants. This is carried out using high-level DFT electronic structure calculations to understand heterogeneous chemical pathways of Hg and CO2 as active materials. The effects of temperature on the adsorption ability of HgCl2 on CaO (001) with 2-D graphene surface support were calculated using DFT. Finally, the auxiliary aspect of this research is to investigate theoretically how carbon dioxide from flue gases of coal-fired power plants is adsorbed on an inorganic g- CaO surface in the presence of water vapor. The research uses a fundamental science-based approach to understand the environmental problems caused by coal-fired energy production and provides solutions to the power generation industry for emissions reductions.
2 Methodology
All calculations were performed using DFT with a B3LYP functional19) with computational cost lowered by the use of highly localized grid representations wherein each electronic wave function is expanded in a localized atom-centered basis set with each basis function defined analytically. The DFT approach can be used to calculate structural properties that are typically within 0.05 Å and 1-2° of experiment. The overall adsorption and reaction energies are typically within 5-7 kcal/mol of experimental values, and spectroscopic analyses are within a few percent of experimental data.6-8) For cluster geometries, spin-unrestricted calculations were carried out with a 6- 31G* valence double-zeta polarized basis set available. Charge densities were analyzed by the Mulliken method.20) For open-shell molecular radicals, the unrestricted formalism was used. The present level of calculation, DFT (UB3LYP)/6-31G*, is known to produce reasonable results 21) for bond lengths, bond angles, and bond energies for a wide range of molecules. The Hg relativistic corrections can be calculated,22) for the relativistic electron density near the nucleus that may be simulated using a relativistic effective core potential. It offers an adequate description of molecules containing Hg wherein the relativistic behavior of the inner electrons cannot be ignored because the standard Schrödinger equation is inadequate for these compounds. The computations were carried out using the ab initio quantum chemistry package, General Atomic and Molecular Electronic Structure System(GAMESS) code, available from Mark Gordons22) Quantum Theory group at Ames laboratory/Iowa State University. The dispersion interaction between two quantum mechanical molecules comes from the general formula for the second-order term in intermolecular perturbation theory. A formula for the dispersion interaction between two molecules based on this functional form model was previously derived and implemented in the GAMESS package.
The Gibbs free energy for the equilibrium constant is calculated using:
where the ΔGads is the change of Gibbs free energy for the adsorption process, the ΔEads of mercury-containing species on the modeled structures, investigated with generalized-gradient approximation GGA/B3LYP methods were calculated through the following equation:
where ΔEads is the adsorption energy, EHgCl2+surface represents the energy of the adsorbate HgCl2-surface, EHgCl2 is the energy of the gas phase mercury-containing species, and Esurface is the energy of the isolated CaO or g-CaO surface model, in kcal/mol. The ΔE0 is the zeropoint energy change during the adsorption process, and ΔSvib, ΔStrans,rot are the vibrational and translational, rotational entropy changes of adsorption in kcal/mol K. T is the temperature in K. k is Boltzmann's constant and the pressure terms cancel out because the pressure is constant in this adsorption system.16,17) The equilibrium constants(Keq) is given as
R is the ideal gas constant in kcal/mol K. The clean perfect surface modeled by a 5 × 5 × 2 CaO (001) structure with and without graphene as 2D support was allowed to relax.
Electronic structure descriptors have been computed to analyze the geometrical and electronic changes such as the ΔG. The above equations were used to obtain the Gibbs free energy and the equilibrium constant for different adsorption scenarios.27-33) Measuring the Gibbs free energy can assess the stability of CO2 in g-CaO with water. A number of quantum mechanical continuum solvation models were developed for this purpose.23,24) These originated from the Onsager continuum model,24) and were formulated by Tomasi et al.23,25,26,27) as the polarizable continuum model(PCM) that is a commonly used method in computational chemistry to model solvation effects. If it were necessary to consider each solvent molecule as a separate molecule, the computational cost of modeling a solvent-mediated chemical reaction would grow prohibitively high. Modeling the solvent as a polarizable continuum, and not as individual molecules, makes ab initio computation feasible, surrounding the solute molecules outside of a molecular cavity.
GaussSum and wxMacMolPlt parses the output files of GAMESS calculations to extract useful information. GaussSum28) tracks the progress of the SCF convergence and geometry optimization, plots the density of states spectrum and the partial density of states, in the case of groups of atoms. The software is written by Noel O'Boyle and is available free under the GNU Public License. wxMacMolPlt29) is a cross-platform gui for preparing, submitting and visualizing input and output for the GAMESS quantum chemistry package its features include a graphical molecule builder, GAMESS input generation, animation of output and visualization of molecules, normal modes, orbitals and other properties.
DFT performed with GGA and the local-density approximation( LDA) of the electronic exchange correlation energy has become the ab initio method of choice for modeling and characterization for mercury and carbon capture materials. Standard DFT-LDA and DFT-GGA methods are known to describe with precision and affordability a wide spectrum of interactions in bulk crystals and surfaces; however, computational studies on the accuracy of DFT in reproducing Hg and CO2-sorbent forces are surprisingly scarce in the literature. In view of the ubiquity of DFT methods, it is therefore crucial to start filling this knowledge gap while putting a special emphasis on the underlying physics. Computational benchmark studies on Hg and CO2-sorbent interactions, however, are technically intricate and conceptually difficult since most Hg and carbon capture materials have structural motifs that are large in size. In particular, genuinely accurate but computationally very intensive quantum chemistry approaches like MP2 and CCSD(T) can deal efficiently only with small systems composed of up to a few tens of atoms, whereas DFT can be used for much larger systems. DFT and MP2 methods qualitatively provide similar results, with hybrid DFT and MP2 in almost quantitative agreement.30)
3 Results and Discussions
3.1 Electronic properties of HgCl2 adsorption on CaO with and without graphene support
HgCl2 exists not as a salt composed of discrete ions, but rather is composed of linear triatomic molecules. In the crystal, each mercury atom is bonded to two close chloride ligands with Hg-Cl distance of 2.38 Å; six more chlorides are more distant at 3.38 Å, as shown in Fig. 1. The distances between mercury and oxygen on the 5 × 5 × 2 CaO surface is 2.344 Å for Hg(+2) at the present calculation levels; the chlorine atom has a bond length of 3.258 Å to the surface with calcium. In both Fig. 1(b) and Fig. 1(d) the movement of the oxygen out of the flat surface makes it more favorable for strong interactions with mercury so that the Hg is much closer to the surface at 2.344 Å for HgCl2 on CaO and 2.441 Å for HgCl2 on g-CaO. Graphene an allotrope of carbon in the structure of a plane of sp2-bonded atoms with a bond length of 0.142 nanometers sandwiched in between the CaO.
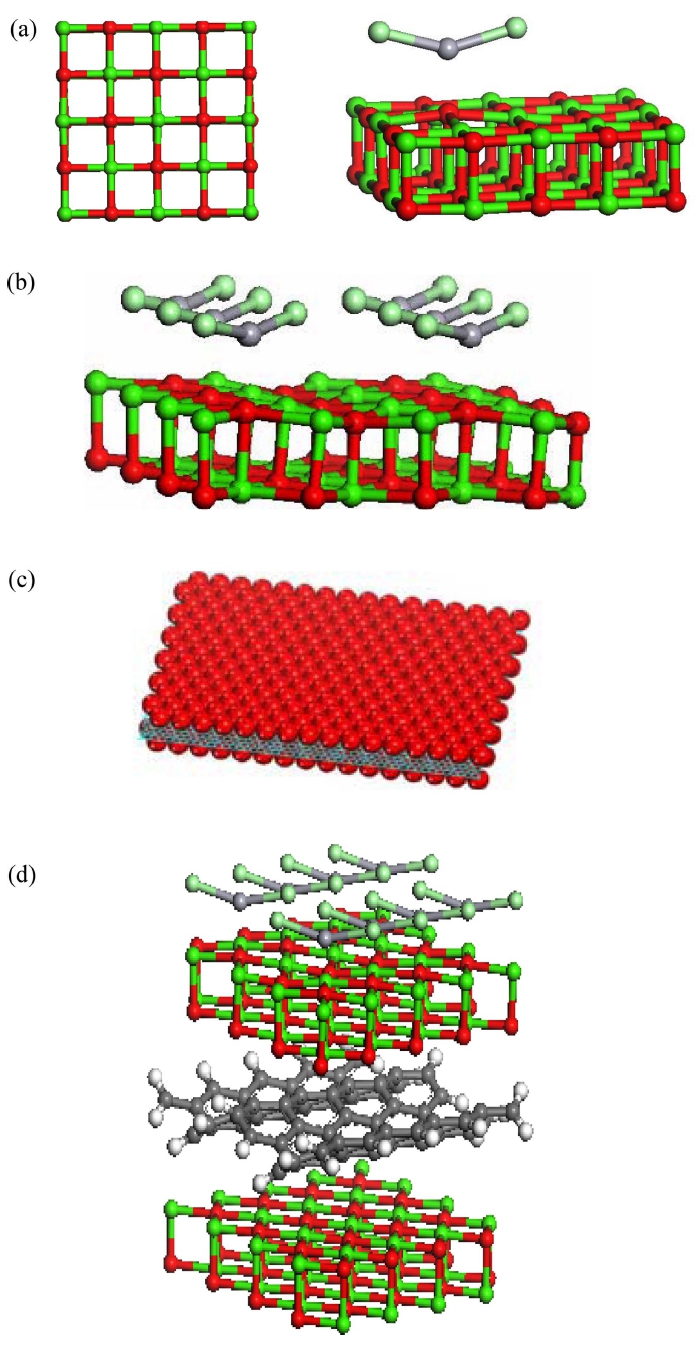
Fig. 1.
Optimized geometry of (a) the finite 5 × 5 × 2 CaO (001) at low concentration, (b) HgCl2 adsorbed on the CaO (001) wherein each chlorine atom gravitated to calcium sites at high concentration (c) Sandwich-like model: graphene serves as a template for the creation of a metal oxide/graphene/metal oxide sandwich-like structure. (d) HgCl2 captured on a CaO and graphene composite at high concentration. Green color depicts Calcium atoms; light green is Chlorine, red is Oxygen, and gray is Mercury. The CO2 system can be modeled simply by replacing the HgCl2 with CO2 molecules.
The adsorption energies of HgCl2 on the modeled structure is −30.28 kcal/mol at low concentration(lCaO); −24.24 kcal/mol at high concentration(hCaO) and −22.80 kcal/mol at high concentration with graphene support(hg- CaO) obtained with B3LYP methods through equation (2). The relaxation effect plays an important role in the HgCl2 adsorption to move the adsorption energy into the chemisorption range. The oxidized mercury has a strong adsorption energy value on the 5 × 5 × 2 slab. The result suggests that CaO will be a good adsorbent for the reactive oxidized mercury species. The adsorption energies of HgCl2 on the CaO surfaces fall within the chemisorption range. The chlorine atoms released from HCl or Cl2 in the coal combustion flue gas system greatly enhanced the adsorption capability according to these predicted results.31)
Although absolute values of Mulliken and the Hirshfeld populations have little physical meaning, their relative values can be useful. For example, positive and negative values of bond population mean that the atoms are bonded or antibonded, respectively. A large positive value indicates that the bond is largely covalent, whereas there is no interaction between the two atoms if the bond population is close to zero. The atomic charges for a fixed clean periodic CaO slab and CaO slab reacted with HgCl2 were used to obtain the final Mulliken and Hirshfeld charges. The Mulliken charge of the mercury atom in HgCl2 on the surface is changed by −0.069, −0.089, −0.077 and the chlorine is negatively charged to −0.145, −0.075, −0.070 for lCaO, hCaO and hg-CaO configurations respectively. On the CaO surface, the final atomic charges of the nearby calcium atoms were almost the same after CaO-Hg adsorption while all charges on nearby oxygen atoms were changed positively after CaOmercury species were formed. The Hirshfeld charges of the nearby calcium atoms were changed by −0.098, −0.098, −0.097 and −0.097 for the hg-CaO as an example. This is consistent with the Mulliken charge analysis as the same calcium atoms of the surface interact with mercury adsorbates.
The aim is to maximize the practical use of the combined advantages of both graphene and metal oxides as active materials for improving the HgCl2 storage. The Calculation of the binding energies of g-CaO structures was elucidated with the following formula:
where ΔEg-CaO is the binding energy of CaO layer with the graphene layer together, Eg-CaO represents the energy of the CaO-graphene composite, ECaO and Eg are the energy of the CaO and graphene layers in kcal/mol. In a composite, graphene provides chemical functionality and compatibility to allow easy processing of metal oxides in the composite. The metal oxide component mainly provides high capacity depending on its structure, size and crystallinity. The resultant composite has a −5.04 kcal/ mol binding energy that is not merely the sum of the individual components, but rather a new material with new functionalities and properties. From the view point of structure, on one hand, CaO anchored or dispersed on graphene can suppress the agglomeration of graphene, which can be accessed by considering now the graphenegraphene interlayer binding energy, ΔEg/g of the bilayer graphene system. The ΔEg/g was found by calculating the total energy of a single graphene layer, and subtracting that from the bilayer calculation, the result is that the ΔEg-CaO is significantly less than ΔEg/g which has a value of −0.42 kcal/mol, supporting the claim that agglomeration with restacking is diminished in the presence of CaO. Also graphene increases the available surface area, leading to high chemical activity. On the other hand, the binding energy of CaO to itself in the presence of graphene is greater than the binding energy of CaO with itself without graphene by 18 kcal/mol which indicates that graphene can act as a support of metal oxides because it can induce the nucleation growth along with the formation of metal oxide nano/microstructures with uniform dispersion and perhaps controlled morphology also on its surface with high chemical functionality. The final metal oxide-anchored graphene and the graphenesupported metal oxide can form a perfect integrated structure with a developed system. The synergistic effects often occurrs on the graphene/metal oxide composites because of size effects and interfacial interactions. Graphene act as a 2D support for uniformly anchoring or dispersing metal oxides with well-defined sizes, shapes and crystallinity; (2) metal oxides suppressing the restacking of graphene; (3) graphene can induce the nucleation growth of metal oxide nano/microstructures and suppress its agglomeration.
Fig. 2, above, shows HOMO(the highest occupied molecular orbital) molecular orbital for the fully optimized 5 × 5 CaO cluster model. For Fig. 2(a) the bare CaO with polarized HOMO at the boundary ends may lead to attraction with adjacent CaO within the vicinity that eventually may result to agglomeration; the edge oxygen atoms have a high energy state and π* molecular orbitals which are formed from 4s(Ca) and 2p(O) atomic orbitals. In Fig. 2(b) the g-CaO sheet without an externally applied electric field is presented, with delocalized HOMO and a localized LUMO(the lowest unoccupied molecular orbital) at the center acts an electrophile to attract electrons from CaO layers in effect the CaO layer is charged less negatively. Polarized HOMO at the boundary ends of the CaO have vanished that can prevent CaO agglomeration. Fig. 2(c) depicts an externally applied field in the g-CaO sheet with polarized HOMO at the boundary ends and delocalized LUMO acts a nucleophile to donate electrons to CaO layers that in effect the CaO layer is more negatively charged. The charged surface is manifested in a darker green color compared to Fig. 2(b). that may lead to an enhanced interaction with HgCl2 and CO2 as discussed in the next. All adsorptions are electronically harmless processes and venial impacts on the energy gap were observed.
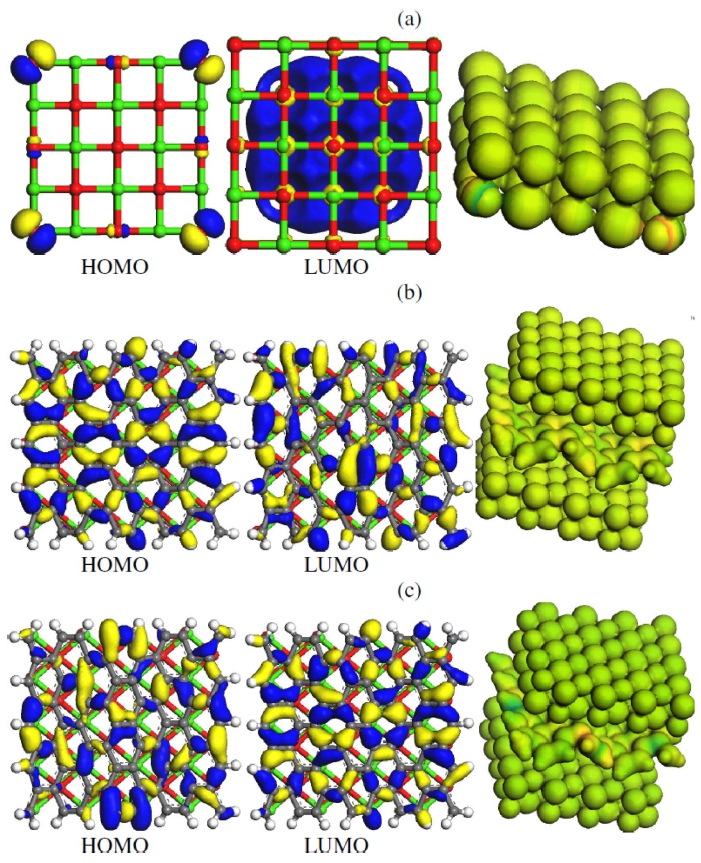
Fig. 2.
HOMO-LUMO surface plots of (a) bare CaO with out an electric field (b) CaO+ graphene composite without an electric field (c) CaO+ graphene composite in the presence of an electric field. Green color depicts Calcium atoms and red is Oxygen.
The effect of temperature on the equilibrium constants for the adsorption of mercury containing species on the CaO (001) surface was investigated with B3LYP calculations. Fig. 3(a) shows that the binding energy increases as the magnitude of the externally applied electric field increases although, it can be noticed that the CaO + graphene composite shown in black increases rapidly compared to the unsupported one; an advantage of graphene/metal oxide composites as advanced materials in HgCl2 capture as seen in the HOMO-LUMO surface plots. Due to the enhanced features of HgCl2 on a CaO (001) with graphene, the predicted equilibrium constant for adsorption of as a function of temperature at high concentration was calculated with and without the in- fluence of an externally applied electric field. Fig. 3(b) shows effect of temperature on the equilibrium constants for the adsorption of HgCl2 in the range of 250-600 K and was generated by using the atomistic thermodynamic calculations described earlier. Temperature has a strong effect on the equilibrium constant for HgCl2 adsorption and the equilibrium constant has a steep change for HgCl2 adsorption. The equilibrium constants decrease as the temperature increases with or without the application of an external electric field and the adsorption of HgCl2 on CaO (001) surface is more favored at low temperatures. In the presence of a weak field of 5.92 kcal/mol Å it can be noticed that there is no significant change in the equilibrium constant however, at 17.77 kcal/mol Å a noticeable increase can be noted already. This is in good agreement with trends for exothermic systems.32-47)However, the small equilibrium constants of HgCl2 at high temperatures means that oxidized forms are not effectively adsorbed under these conditions.
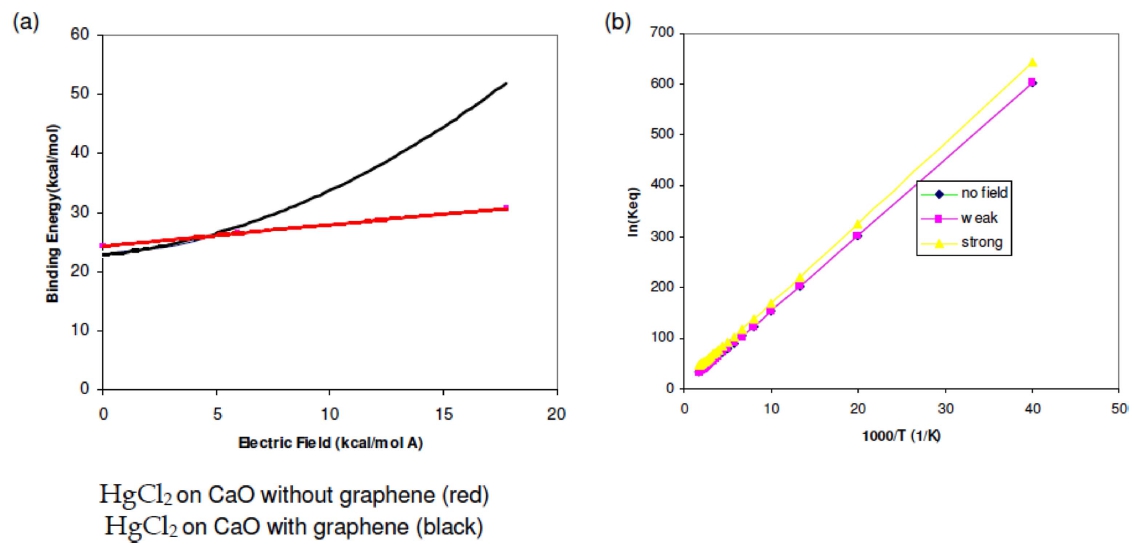
Fig. 3.
The (a) binding energy of HgCl2 on CaO with and without graphene reinforcement versus an externally applied electric field. The (b) predicted equilibrium constant for adsorption of HgCl2 on a CaO (001) with graphene as a function of temperature at high concentration calculated with the B3LYP functional with and without the influence of an externally applied electric field. Weak field is 5.92 kcal/mol Å and strong field is 17.77 kcal/mol Å
One recovers the used adsorbents despite of the high adsorption energies at high heating rates, the desorption is kinetically favored. Although other effects connected with the physical form of the specie deposited must be considered. The desorption can be dependent on the dielectric and surface properties, electric field strength. The response of the specie to changes in surface properties upon rapid heating might result in favorable modifications of the surface charging effects, and other factors can also assist the desorption under rapid heating conditions. In general, the use of high electric-field gradients and heating can readily cause desorption from the surface.
3.2 Electronic properties of CO2 adsorption on g-CaO support with and without the presence of water vapor
Carbon dioxide is chosen as the reference gas to calculate global warming potentials for other greenhouse gases because of its large contribution. Simple carbonation (Eq. 5) and hydration(Eq. 6) reactions were used namely:
The change of enthalpy for calcium carbonate CaCO3 is −42.49 kcal/mol and it is a strongly exothermic reaction. The calculated enthalpy change was −55.87 kcal/mol, which yields a 24 % error compared to the experimental value. This confirmed one molecule of calcium carbonate could not lead to reliable adsorption results because it exists in a crystal structure. Therefore, the surface adsorption method was applied and a 5 × 5 × 2 CaO surface.
The distance between carbon of the carbon dioxide molecule and oxygen on the g-CaO surface is 1.37 Å after optimization. The bond lengths between carbon and the oxygen atoms of CO2 molecule have changed from 1.51 Å to 1.25 Å when the double bonds are broken. In the calcite(CaCO3) structure, the default bond lengths between carbon and oxygen are all equally 1.28 Å. As the g-CaO surface is relaxed, the optimized structure shows similarity to the calcite geometry. When the CO2 adsorbs on the g-CaO surface in the presence of water vapor in the PCM model, it has the same geometry as the CO2-only adsorption model. This indicates that water vapor in the PCM formalism does not affect the optimized CO2 adsorption geometry in forming the calcite structure. The adsorption energies were calculated using optimized structures obtained from the previous calculations in section 3.1 by replacing the HgCl2 with CO2 molecules where they are defined by
The adsorption energies of CO2 with and without water on the 5 × 5 × 2 structure were −23.82 and −40.31 kcal/ mol, respectively. The adsorption energy for strong CO2 is in the chemical adsorption range in agreement with literature.38-39) The water vapor enhances the adsorption ability of CO2 on the relaxed g-CaO surface as it increases the adsorption energy by ~ −16 kcal/mol. The water vapor leads to more favorable CO2 adsorption on the relaxed CaO/gaphene here. The strong hydration effect is in agreement with experimental results. Lime-based sorbents are used for fuel- and flue-gas capture, thereby representing an economic and effective way to reduce CO2 emissions. Their use involves cyclic carbonation/calcination, which results in a significant conversion reduction with increasing number of cycles. To reactivate spent CaO, vapor phase hydration is typically performed. Steam was attributed to improving carbonation performance on graphene/CaO surfaces. The adsorptions are electronically harmless as with the HgCl2 adsorption on CaO with and without the graphene support.
The ΔG of carbonation reaction on the model CaO surface is −32.47 kcal/mol at 298.15 K, with only a 5 % error compared to experiment. The ΔG is also the chemical potential that is minimized when a system reaches equilibrium at constant pressure and temperature. Its derivative with respect to the reaction coordinate of the system vanishes at the equilibrium point. As such, it is a convenient criterion of spontaneity for processes with constant pressure and temperature. The results of CO2 adsorption were compared with the Gibbs energy for CO2 adsorption with water vapor, the hydration effect contributed to much more favorable adsorption at −39.17 kcal/mol. The high adsorbability of CO2 with a water molecule using the PCM model on the relaxed CaO surface is valuable for real flue gas systems. Most CO2 exists in the presence of water vapor. However, CO2 adsorption without a water molecule on the relaxed CaO surface is more favorable at high temperatures. A water molecule in the liquid form, which is the condition at lower temperatures of the flue gases, would lead to more favorable CO2 adsorption on calcium oxide surface. Regardless, the ability for adsorbing CO2 in the presence, or absence, of a water molecule on the CaO/graphene slab is chemisorptions at all temperature ranges. Desorption is suggested to be achieved also using high electric-field gradients.
4 Conclusions
The effect of temperature on the equilibrium constant for the adsorption of HgCl2 species on 5 × 5 × 2 g-CaO surface was investigated by using the GGA/B3LYP calculations. Results show that graphene is a support for uniform dispersion of CaO; the metal oxide suppresses the re-stacking of graphene and graphene suppresses the volume change and agglomeration of CaO as supported by HOMO-LUMO surface plots. The compound, HgCl2 was found to be stable on the surface; it does not desorb or return to the reactant species. The equilibrium constants for mercury adsorption on CaO surfaces with and without the presence of an externally applied electric field are affected by temperature. From the trends, HgCl2 adsorption on g-CaO surfaces is favorable at low flue gas temperatures. Carbon dioxide adsorption on the g-CaO slab surface led to formation of a calcium carbonate structure through the carbonation reaction. There was enhanced CO2 adsorption ability in the presence of water vapor. Results show that water molecules mediated through PCM model leads to enhanced CO2 adsorption on the relaxed g-CaO surface through the hydration effect. Understanding the mechanism associated with Hg and CO2 adsorption on g-CaO will help to optimize the conditions to limit emissions from coal-fired power plant processes.
