

1.서 론
산화물 지지체에 지지된 귀금속 촉매입자는 가장 대표 적인 불균일 촉매의 형태이다.1-3) 이러한 귀금속/산화물 촉매에 존재하는 귀금속-산화물의 계면은 다양한 촉매 반 응에 대한 활성점으로 작용한다고 알려져 있다.4-13) 최근 금(Au), 백금(Pt), 팔라듐(Pd) 등 대표적 귀금속 촉매 소 재의 가격 상승이 지속되면서 금속 나노입자의 크기를 줄 여 1 nm 미만의 클러스터나 단원자(single-atom)들을 촉매 로 활용하여 소재 활용량 당 활성을 높이고 귀금속 소재 의 효율적 사용을 꾀하려는 시도들이 나타나고 있다.10,14,15) 이에 따라, 귀금속/산화물 촉매 표면에 존재하는 금속-산 화물 계면의 상대적인 넓이 분율이 증가하면서, 금속-산 화물 계면의 촉매 특성을 조정하거나 최적화하는 것이 불균일 촉매 디자인을 위한 새로운 방법으로 자리잡고 있다.16-21)
세륨산화물(CeO2, Ceria)은 산소 공공의 형성이 용이해 산화반응을 촉진할 수 있어 TiO2와 함께 귀금속/산화물 계열 촉매에 널리 사용되는 가장 대표적인 지지체 소재 이다.8,11-13,19) 금 나노입자를 CeO2에 결합한 촉매는 일산 화탄소 산화반응(CO oxidation, 2CO + O2 → 2CO2),8) 수성가스(水性gas) 전환반응(water-gas shift reaction, WGSR, H2O + CO → H2 + CO2)13) 등에 높은 활성을 보인다고 알려져 있으며, 특히 저온에서도 이들 반응에 대한 활 성이 우수하다고 알려져 있다. 일산화탄소 산화반응과 수 성가스 전환반응은 메탄 개질을 이용한 수소 생산과정 에 깊이 관여하고 있으며,8,20) 특히 이들의 저온 활성을 높이는 것이 전체 수소 생산과정의 효율을 높이는데 꼭 필요하다.
일반적으로 금은 나노입자의 경우에도 산소에 대한 친 화력이 일산화탄소에 비해 상대적으로 매우 낮고12) 따 라서 일산화탄소가 존재하는 반응환경에서는 일산화탄소 가 금 나노입자의 표면의 대부분을 덮는 현상이 발생한 다.8,22) 따라서 금-산화물 계열 촉매의 산화반응에 대한 우수한 활성은 금과 산화물의 계면에서 일어나는 산소 공급에 의해 일어난다는 것이 일반적인 사실로 인정되 고 있다.8,9,11-13,22) 그러나 금-산화물의 계면이 WGSR와 같이 물 분자를 산소 공급원으로 활용하는 반응이나 선 택적 일산화탄소 산화 반응(preferential oxidation of CO, PROX, 2CO + H2 + O2 → H2 + 2CO2)과 같이 두 종류의 환원성 물질이 함께 공급되는 반응에 대해 어떠 한 방식으로 촉매 반응에 관여하는지는 명확하지 않다. 또한, 이와 같은 촉매 반응들의 활성은 반응 가스의 공 급량(flow rate), 반응 가스내 반응원의 부분압 등, 환경 적 요소에 의해서도 좌우되기 때문에 금속-산화물 계면 이 가지는 근원적 활성에 대한 연구의 필요성이 높다.
본 논문에서는 전자밀도함수이론(density functional theory, DFT) 계산을 연구 방법으로 활용하여 CeO2(111) 표면에 존재하는 결함(defect)에 금 나노입자를 지지하여 Au/CeO2(111) 촉매 시스템을 설계하고 Au-CeO2 계면이 물 분자에 대해 나타내는 활성을 분석하였다. 특히, CeO2 표면에서 나타나는 물 흡착 및 활성에 대한 본 그룹의 이전 연구결과와 비교하여 Au-CeO2 계면의 특성을 해 석하고 Au/CeO2 촉매 시스템의 촉매 활성을 통합적으 로 이해할 수 있게 하였다.
2. 연구방법
본 연구에서는 DFT 계산 수행을 위해 vienna ab-initio simulation package (VASP)23) 프로그램을 사용하였다. Hubbard에 의해 제시되고 일반적으로 DFT+U라고 알려 져 있는24) 전자 분포 포텐셜 조정 방법을 Ce 이온들의 localized f 전자의 상태를 묘사하였다. 이때 Ueff는 5 eV 로 설정하였다. 또한, Fernández-Torre의 연구 결과를 바 탕으로25) non-local correlation이 포함된 vdW-corrected vdW+DF26,27) 방법과 함께 optB86b exchange functional 을 활용하여 vdW 힘의 영향을 고려하였다.25) 400 eV의 에너지를 가지는 평면파 basis set과 projector augmented wave method를28) 사용하여 최외곽 전자의 분포와 이들과 양이온으로 묘사된 원자 중심부의 상호작용을 해석하였 다. 연구에 사용한 Au-CeO2 시스템의 크기가 크다는 것 을 감안하여 brillouin zone sampling은 gamma point만을 이용하였다. 원자구조(geometry) 및 전자구조 최적화에 대 한 수렴 판단은 각각 0.01 eV/A와 10−4 eV의 기준을 활 용하였다. 페르미 레벨 근방의 전자들에 대한 수렴을 촉진 하기 위해 0.1 eV의 온도 변위를 통한 gaussian smearing function을 적용하였다.
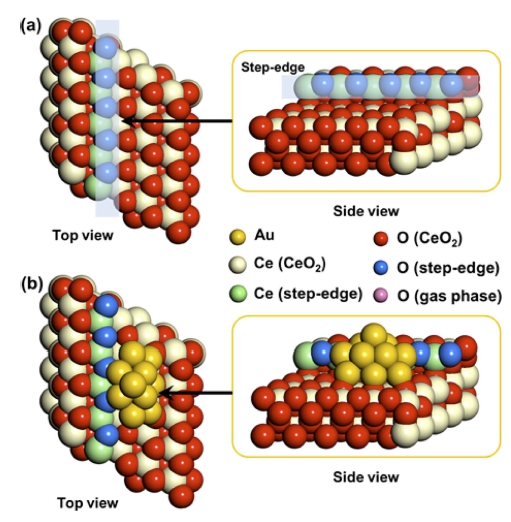
6개의 원자층으로 이루어진 5 × 5 CeO2(111) slab 구 조의 표면에 2/5 monolayer에 해당하는 Ce10O20 strip을 추가하여 표면 step이 존재하는 CeO2(111) supercell을 구성하였다(Fig. 1).5) 이때 표면에 존재하는 step-edge에 12개의 원자를 가지는 Au 나노입자를 부착하여 전체 촉 매 시스템을 완성하였다. 이와 같은 촉매 모델들의 신 뢰성은 이전 연구 결과를 통해 증명된 바 있다.5)
3. 결과 및 고찰
Fig. 1은 계산에 사용한 Au/CeO2시스템의 구조를 보 여준다. 먼저 WGSR에서 필수적으로 일어나는 물 분자 의 흡착과 활성화 지점을 찾기 위해 Au 나노입자의 표 면과 계면에 대한 흡착 에너지(Ebind) 계산을 수행하였 다. 일반적으로 Au 나노입자는 약 -1.0 eV 내외의 Ebind 를 가지고 일산화탄소를 흡착한다.8,22) 또한 DFT를 통해 계산한 개별 분자들의 흡착 성향을 분석하고 비교하기 위해서는 흡착 과정에서 일어나는 엔트로피의 감소와 흡 착이 일어난 후 촉매 표면의 configuration 엔트로피 증 가에 의한 추가 기여를 보정하여 깁스 자유에너지를 계 상하여야 한다. 그러나 일산화탄소와 물의 단일 분자 흡 착 에너지의 차이가 매우 크게 되면 두 분자의 흡착 과 정에서 일어나는 각각의 엔트로피 추가 기여에 의한 차 이를 무시할 수 있게 되어 Ebind를 통해 분자들의 흡착 경향을 비교, 분석할 수 있게 된다.
Fig. 2에 나타낸 Au/CeO2에 대한 물 분자의 흡착 지 점과 Ebind를 비교하면, 물 분자가 Au 나노입자의 표면 에 흡착하는 경우 Ebind = -0.32 eV를, Au 나노입자와 CeO2의 계면에 물 분자가 흡착하는 경우 Ebind = -1.55 eV를 나타냄을 알 수 있다. 일산화탄소, 물, 산소 등의 분자들의 표준 엔트로피가 300 K에서 약 0.6 eV 내외임 을 고려해볼 때,8,12) Au 나노입자의 표면은 물 분자를 안 정화시키기 어렵다고 판단할 수 있으나 Au-CeO2 계면 은 물 분자를 흡착하고 고정할 수 있음을 알 수 있다. 또한 Au와 인접한 CeO2 step-edge 부근에 물 분자가 흡 착하는 경우를 6종류 이상 확인하였으나 이들의 Ebind는 바로 인근의 Au-CeO2 계면의 Ebind = -1.55 eV 대비 절 반 가량에(-0.64 eV ~ -0.78 eV) 그쳐, 물 분자의 활성화 에 끼치는 영향이 적을 것으로 판단된다. WGSR과 같 이 일산화탄소가 함께 존재하는 경우에서는 Au 나노입 자의 표면은 대부분 상대적으로 강한 Ebind를 가지는 일 산화탄소를 선호할 것으로 생각된다. 이와 같은 현상은 Ha 등에 의해 수행된 DFT 계산 결과를 통해서 증명된 바 있으며,8) Pt 나노입자에 대해 표면의 CO 흡착 비율 을 변화하면서 수행한 DFT 계산 결과에서도 유사하게 나타난 바 있다.29)
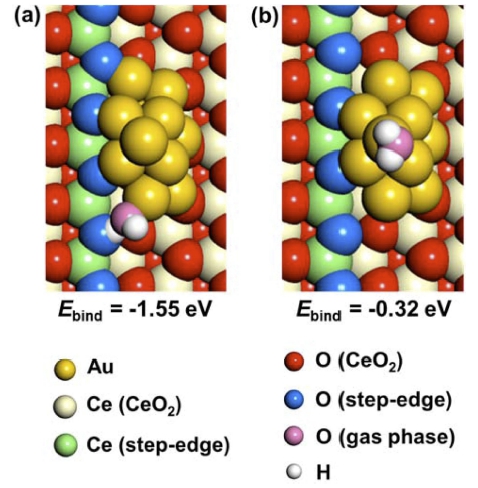
Fig. 2
Geometry and adsorption energy of a water molecule (a) at the Au-CeO2 interface or (b) on the Au nanoparticle of a Au/CeO2 model. The Ebind denotes the DFT-calculated binding energy of water.
Fig. 3(a)는 Au-CeO2 계면에서 강하게 흡착하는 물 분 자의 원자 결합 길이 변화를 나타낸다. 일반적으로 물 분자가 촉매 표면에 흡착되는 경우에는 원자간 결합길 이의 변화가 없는 분자형(molecular adsorption) 흡착과 원 자 결합 길이가 변하는 분리형(dissociative adsorption) 흡 착이 일어날 수 있다고 알려져있다. Fig. 3(a)에 나타낸 구조에서는 물 분자의 H-O-H 결합길이가 원래의 길이 인 1.0 Å30) 내외에서 1.608 Å까지 증가한 것을 알 수 있 으며, 이를 통해 Au-CeO2 계면에서 물 분자가 흡착, 활 성화 된다는 것을 알 수 있다. 특이한 것은 물이 존재하 지 않는 경우에 Au-CeO2 계면 부근에 존재하는 Au 원 자들은 일산화탄소를 강하게 흡착하는데,8) 물이 존재하 는 경우에는 이와 같은 계면 근처의 Au 원자가 물을 상 대적으로 더 강하게 흡착한다는 점이다. Au/CeO2 촉매 에서 일어나는 일반적인 일산화탄소 산화반응에서는 Au- CeO2 계면에서 CeO2가 산소를 직접 공급하면서 Au에 흡착된 일산화탄소의 산화반응이 일어난다고 알려져 있 는데 반해,8) Fig. 2와 3(a)에 나타낸 결과들은 WGSR과 같이 물이 존재하는 환경에서 일어나는 일산화탄소 산 화반응은 Au와 CeO2의 계면에서 활성화되는 물 분자에 의해 산소를 공급받을 수 있다는 것을 보여준다.
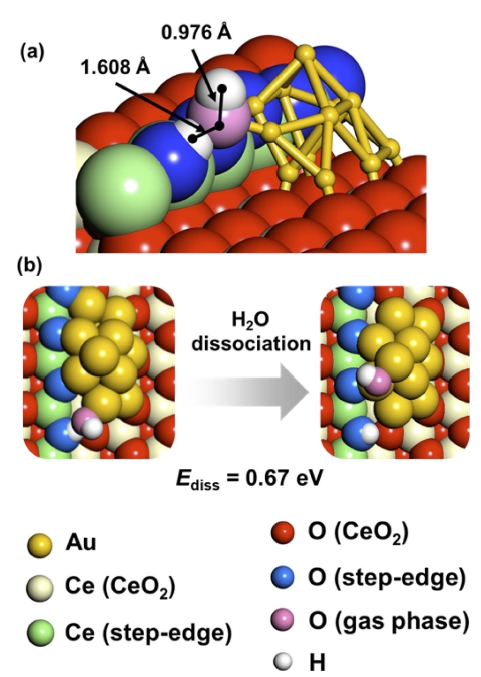
Fig. 3
Water adsorption and dissociation at the Au-CeO2 interface of a Au/CeO2 catalyst model. (a) Elongated O-H bond distance indicates that the adsorbed water mole is activated. (b) Energetics of subsequent water dissociation (Ediss) shows that the dissociation is slightly uphill.
Fig. 3(b)에서는 이와 같이 활성화된 물 분자의 분리 (dissociation)에 필요한 Ediss를 나타내었다. 비록 분리된 –OH와 –H의 위치에 따라 Ediss가 달라질 수 있으나, 물 분자의 분리가 0.67 eV의 에너지 공급을 통해 가능한 것 으로 나타나고 있다. CeO2 표면에서 산소가 공급되는 형태의 일반적인 일산화탄소 산화반응을 가정하고 계산 한 CeO2의 산소 공급 에너지 즉, 산소 공공 형성에너 지(oxygen vacancy formation energy)가 CeO2의 표면의 형태에 따라 대략적으로 1.02 eV ~ 2.48 eV에 이르고,11,12) 본 연구에 사용한 CeO2(111) step edge와 Au/CeO2 구 조에서 계산한 산소 공공 형성에너지가 각각 1.66 eV와 1.02 eV 내외임을 고려하면,5) Au와 CeO2 계면에서 흡착 되는 물이 공급하는 산소가 좀 더 에너지 관점에서 평 이하게 활용될 수 있는 가능성이 있다.
4. 결 론
본 연구에서는 Au 나노입자와 CeO2로 이루어진 촉매 시스템에 대해 vdW 힘이 고려된 전자밀도함수이론 계 산을 활용하여 물의 흡착과 활성화 특성을 분석하였다. Au 나노입자와 CeO2(111) 표면에 존재하는 step-edge로 이루어진 계면은 Au 나노입자의 표면 대비 물 분자에 대한 흡착력이 약 2배 가량 높았으며, 이로 인해 물 분 자를 흡착하여 활성화할 수 있었다. 또한, 계면에 흡착 된 물 분자가 분리되는데 대한 열역학적 구동력이 있음 을 발견하여 Au와 CeO2의 계면이 물 분자에 존재하는 산소를 활용하는 화학반응(예: WGSR)에 대해 산소 공 급지점으로 작용할 수 있음을 확인하였다. 저온에서 Au/ CeO2 촉매에 의해 일어나는 일산화탄소 산화반응이나 WGSR에서 Au 나노입자가 일산화탄소 흡착을 담당하는 것을 고려하면, 이와 같은 Au-CeO2 계면 특성에 대한 분석 결과들은 CeO2가 산소를 공급하는 단순 일산화탄 소 산화반응이나 WGSR와 같이 물에 의해 산소를 공급 받는 일산화탄소 산화반응 모두 Au-CeO2의 계면이 중 요한 역할을 담당한다는 것을 보여준다. 본 연구 결과 는 Au/CeO2 촉매를 활용하는 화학반응의 경우 계면의 역할을 최우선적으로 고려하여 촉매를 분석하고 디자인 하는 것이 효율적일 수 있음을 의미한다.
