

1. Introduction
Since early experimental findings that oxide supported small Au nanoparticles (NPs) catalyze CO oxidation even at or below room temperature,1-2) many efforts have been devoted to understand the chemical factors that bring the exceptional catalytic properties of Au catalysts. However, given the complexity of supported Au NPs, details of the chemical state of the reactive species are still vague. The factors that may make supported Au NPs catalytically active have been considered thus far with the size effect,3-12) electronic interaction between supporting materials and NPs,7,13-19) and the presence of NP-support interface.6,9,18,20-26)
A group of recent reports have attributed the origin of the excellent catalytic activity of supported Au NPs to the fact that they have smaller dimensions than previously anticipated.27-30) Hutchings and co-workers found that very small Au NPs consisting of around 10 atoms each are responsible for the CO oxidation reactivity of iron-oxide-supported Au NPs.27) Based on experimental and theoretical studies, Lee et al. showed that oxidesupported tiny Au6-10 NPs (consisting of 6 to10 atoms) are active for selective propene epoxidation.29) Turner et al. experimentally demonstrated that supported small Au NPs (~1.4 nm) derived from Au55 NP can activate molecular oxygen, leading to moderate activity for selective oxidation of styrene.30) They also indicated that Au NPs with diameters of ~2 nm and above are completely inactive. 30) Moreover, single atom reaction centers or subnanometer sized Au NPs are getting more attentions as a reaction centers for reactions catalyzed by supported Au NPs.31-32)
Given the fact that very small Au NPs are potentially, catalytically active, the importance of low-coordinated surface atoms is gaining increasing credibility. Liu et al. found that a low-coordinated step on the Au (111) surface is reactive for CO oxidation.20) Moreover, comparing CO oxidation activity of the Au closed-packed surface and that of the Au12 NP, Nørskov and co-workers confirmed that a low-coordinated corner of the Au12 NP facilitates the reaction.5) However, in a natural extension of Nørskov’s findings, Illas and co-workers studied adsorption and dissociation energies of O2 on Au25, Au38, Au55, and Au79 NPs and found that the O2 adsorption energy is not a simple function of coordination number or particle size.10) We also found that the activity of a specific reaction center of small unsupported Au NPs for CO oxidation is not just a simple function of the Au-Au coordination number.33)
Other noticeable recent studies have focused on the direct or indirect participation of the Au-support interface for catalytic oxidation.23-25,34-35) These studies suggest an essential role of the Au-oxide interface for O2 binding or oxygen supply for subsequent oxidation of CO bind to Au NP.
However, because heterogeneous catalyst is a composite material itself and we are observing the averaged external properties of comprising materials, even though the interfacial areas or the low-coordinated sites could attribute to the relatively larger portion of the total activity, their combination or synergistic effect should be always considered to understand the origin of the catalytic properties of Au-oxide catalyst systems.
In this aspect, we performed a set of density functional theory (DFT) calculations to provide insights into the origins of catalytic activity of Au NPs. We initially intend to quantitatively study the effects of the size, shape, and coordination number of Au NPs on their O2 affinity, which is a critical step for oxidation reactions catalyzed by metal NPs, especially we discuss the O2 affinity of our studied Au NPs considering the mechanism of catalytic CO oxidation. To focus on such intrinsic factors of Au NPs, we excluded the oxide supports. In this study, we examined the oxygen adsorption on 9 types of small unsupported Au NPs of around 1 nm in size.
2. Computational Details
In accordance with the magic number of atomic construction of small NPs, we prepared the following 6 unsupported Au NPs: Au13-Icosahedrol (Ih), Au13-Cubo- Octahedron (COh), Au19-Octahedron (Oh), Au38-Truncated- Octahedron (TOh), Au55-Ih, and Au55-COh (see Fig. 1). We excluded Au NPs larger than Au55 because the oxygen chemistry of Au79-TOh converges to that of bulk Au surface.10) Experimental and theoretical studies have shown that a planar-to-non-planar turnover occurs in Au NPs with 8 atoms36) or 12 to 14 atoms.37) The exact point for this turnover is still in debate. In order to ensure the consistency in morphology over the studied Au NPs, we studied Au NPs larger than Au13, the minimum Au NP size for which they clearly show 3-dimensional crystalline structure. We additionally considered two experimentally reported small Au NPs, Au19-Truncated-Tetrahedron (TTet) and Au20-Tetrahedron (Tet),38-39) as well as a specifically designed Au25 that comprises one-half of Au38-TOh and is regarded as a representative of supported Au NPs.10,40)
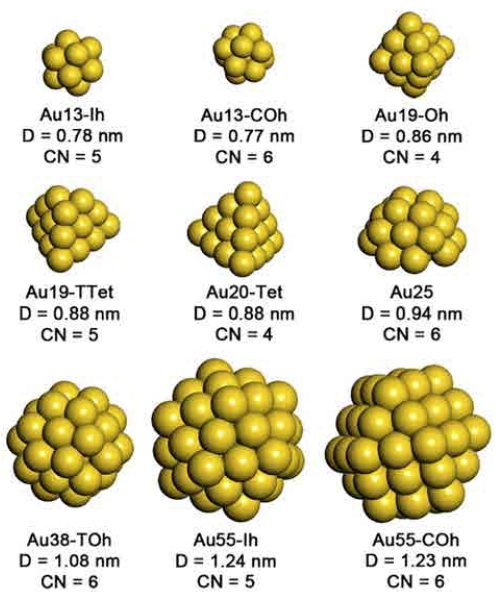
Fig. 1
Morphology and structural information of studied 9 Au nanoparticles. D and CN denote the diameter and the lowest coordination number of NPs.
We performed GGA-level spin-polarized DFT calculations with DMol3 code.41-42) The exchange-correlation energy was functionalized with the PW91,43) PBE,44) and RPBE45) functionals. Roldán et al. showed that energy of adsorption of O2 on gas-phase Au NPs is highly dependent on the exchange-correlation functional.40) They compared the performances of three general GGA-level exchange-correlation functionals, PW91, PBE, and RPBE. The PW91 functional overestimated the oxygen adsorption, whereas PBE and RPBE functionals predict rather weak oxygen-Au NP interaction.40) Generally, the oxygen binding energy calculated with the PW91 functional is rather closer to the LDA values and the RPBE functional relatively accurately describes the surface chemistry of transition metals.40,45-46) The Kohn-Sham equation was expanded in a double-numeric quality basis set with polarization functions (DNP). The orbital cutoff range was 5.0 Å. The DFT semi-core pseudo potential47) was used to treat the core electrons of Au atoms. We also used a Fermi smearing method with a window size of 0.007 hartree (1 hartree = 27.2114 eV). The energy, force, and displacement convergence criteria were set to 10−5 hartree, 0.002 hartree/Å, and 0.005 Å, respectively.
Prior to evaluating O2 and CO adsorption on Au NPs, their structure was fully optimized. Fig. 1 shows the morphology and structural information of the studied Au NPs. The diameters of the Au NPs, calculated using the particle volumes, lie between 0.77 nm (Au13-Ih) and 1.24 nm (Au55-Ih). The coordination numbers of the most under-coordinated surface atoms of the NPs vary between 4 and 6.
To date, depending on the type of reaction intermediate, an association mechanism (O-O-CO, four-center intermediate) 5,20,48) and a carbonate mediating mechanism (OCOO, carbonate-like intermediate)49-51) have been proposed as a reaction mechanism of CO oxidation catalyzed by small NPs. However, in both cases, moderate O2 adsorption is a minimum requirement for CO oxidation catalyzed by small metal NPs.49) For sound operation of CO oxidation, the energy of O2 adsorption should be sufficiently strong to prevent the thermal desorption of the O2 molecule. Since O2 adsorption is an initial step for CO oxidation by metal NPs irrespective of the kind of reaction mechanism, 5,48-49) we postulate that stronger O2 adsorption than its gas phase free energy (−0.63 eV at 298 K and 1 atm) would maximize the population of adsorbed O2 molecules that participate in further reaction processes leading to the maximum activity. However, excessively strong O2 adsorption is not favorable, as it may lead to unwanted permanent oxidation of NPs.
Because the under-coordinated surface atoms where the excess electrons are localized are the potential candidates for O2 adsorption, we first examined O2 adsorption on the edges and vertices of Au NPs.
3. Results and Discussion
Fig. 2 and Table 1 show the strongest O2 adsorption geometry and corresponding energy of adsorption of studied NPs. Trends in O2 adsorption energy in Table 2 substantially shows that the PW91 predicts the strongest O2 adsorption whereas the RPBE predicts the weakest O2 adsorption energy. We found, however, that the O2 adsorption geometry is not dependent to the choice of the GGA-functionals. Irrespective of the GGA-functionals, the O-O bond distance of the adsorbed O2 molecule was substantially elongated relative to that of the gas phase O2 molecule (1.24 Å) being the clear evidence of the electron flow from the Au NP to the adsorbed O2 molecule and the presence of the chemical interaction between them. Even the RPBE functional is the most recent and relatively accurately describes the oxygen chemistry of transition metal surfaces. Table 1 shows that the RPBE functional mostly produces positive O2 adsorption energy on Au NPs larger than the Au13. As the elongated O-O bond distance is the evidence of the chemical interaction between the O2 molecule and the Au NP (see Fig. 2), we chose the PBE functional that produces weak but negative O2 adsorption over studied Au NPs for further discussion.
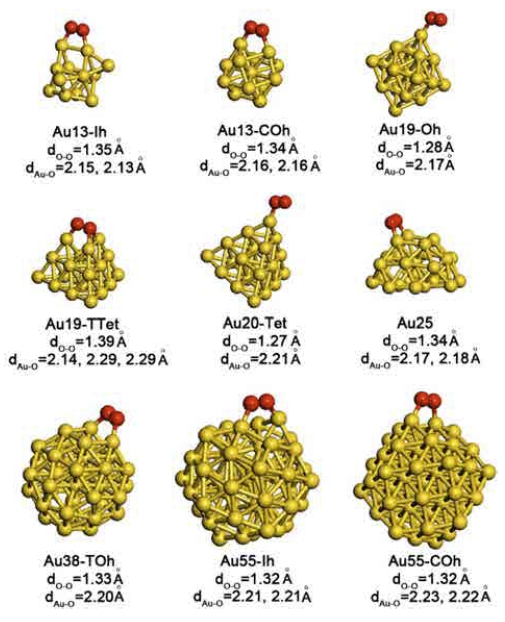
Fig. 2
Most favorable O2 adsorption geometry of studied Au NPs. dO-O and dAu-O denotes O-O bond length of the adsorbed O2 molecule and Au-O distance.
As we anticipated, the most favorable O2 adsorption occurs at the corners and vertices of NPs (see Fig. 2). Among the 9 studied Au NPs, the Au13-Ih ( = −1.96 eV), for which the O2 adsorption causes a severe structural change, interacts most strongly with an O2 molecule (see Fig. 3), whereas the other studied Au NPs weakly bind an O2 molecule. We believe that the low O2 binding energies of other Au NPs for example Au19-TTet and Au55-Ih are accompanied by the extremely short life span of the adsorbed O2 molecule. Therefore, further CO oxidation reaction would not occur or CO2 production would be very low. We postulate that our unsupported Au NPs, except for the Au13-Ih, are by themselves not good catalysts for CO oxidation.
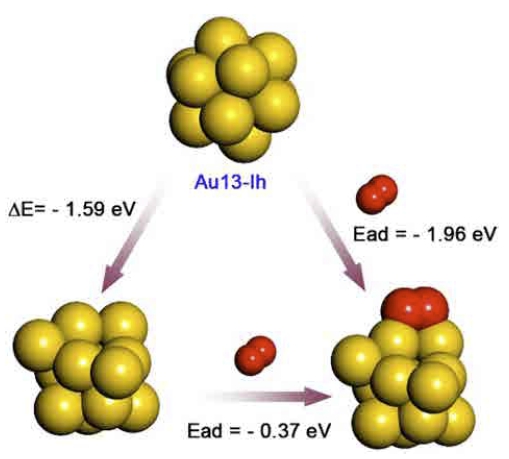
Fig. 3
O2 adsorption on the Au13-Ih. The adsorption-driven structural change occurs during O2 adsorption; more than 80 % of total adsorption energy is attributed to the energy gained by the structural change of the Au13-Ih.
However, it should be noted that almost 80 % of O2 adsorption energy of the Au13-Ih is attributed to the energy gained by the adsorption-driven structural change. Such structural evolution has also been observed in our previous study of CO oxidation by the Ag13-Ih.28) Spontaneous structural evolution is energetically plausible because the final structure of the Au13 NP generated after O2 adsorption is more stable than the Au13-Ih, by as much as 1.60 eV (Fig. 3). An O2 molecule draws electrons from the contacting Au atoms and leads to positively charged Au atoms. Such electron redistribution causes an electrostatic repulsion between the positively charged Au atoms, resulting in a structural evolution of the Au13-Ih NP. However, if we exclude the energy gained by the structural change of the Au13-Ih, the energy of O2 adsorption is decreased to −0.36 eV (Fig. 3). This confirms that the strong O2 adsorption of the Au13-Ih is mostly due to the adsorption-driven structural change.
We introduced the Au13-Ih and the Au13-COh in order to ensure the consistency in morphology over the studied Au NPs. However, these are not the ground state structure of the Au13 NP. The disordered form of the Au13-Ih which is shown in Fig. 3 is energetically more stable as much as −1.60 eV than the Au13-Ih and −0.97 eV than the Au13-COh. Oviedo and Palmer reported that there are many structural isomers of Au13 NPs and pointed out that the disordered Au13 is energetically more stable than crystalline ones.52) Our present results also confirm that the disordered form of the Au13 NPs is energetically more stable than crystalline Au13 NPs. Noticeably, Oviedo and Palmer showed that the energy gaps between the structural isomers of the Au13 NP are very narrow.52) Therefore, even though we used the Au13-Ih as an initial structure, spontaneous structural transformations between the isomers would be observable even at room temperature. We believe that the structural fluxionality of the Au13 NP would contribute to the catalytic activity.
The fact that the structural fluxionality of very small NPs may advantageous to O2 adsorption was proposed by Lopez and Nørskov on unsupported Au10 NP,53) Yoon et al. on MgO supported Au8 NP,19) Remediakis et al. on rutile TiO2 supported Au10 NP.22) Considering our results on the Au13-Ih and reported ones on CO oxidation by small Au NPs,19,22,53) such adsorption-driven structural change generally occurs on very small Au NPs irrespective of the presence of supporting oxides.
Our results show that although the coordination number, size, or morphology of crystalline unsupported Au NPs affects on their O2 adsorption chemistry, there is no clear quantitative relationship between the energy of O2 adsorption and the coordination number, size, or morphology of Au NPs composed of 13 to 55 atoms (the Au13-Ih is an exception). None of these factors can dominantly control the catalytic activity of small Au NPs. In fact, the Au55-Ih and the Au19-Oh slightly strongly bind an O2 molecule than the Au55-COh and the Au19-TTet. However, the differences in O2 adsorption energy are not enough to make the former NPs more reactive than the later NPs.
To our best knowledge, previous studies on the relationship between the size, shape and coordination number of small Au NPs and their surface chemistry just reported that some specific Au NPs show better or worse properties than others.3,10,40) Information is fragmentary to deduce the general rule. In the size range of our NPs, except for the Au13-Ih, it is likely not true that the smaller NPs are always more reactive than the larger NPs and that the NPs with the lower-coordinated surface atom ensure higher reactive.
The Au20-Tet (coordination number = 4) that very weakly or not at all binds an O2 molecule irrespective of the GGA-functionals confirms our speculation (Table 1). Li et al. experimentally found that the Au20-Tet has extremely high HOMO-LUMO gap (1.8 eV) and suggested that such high energy gap makes the NP chemically inert.38) We also found that the calculated HOMO-LUMO gap of the Au20-Tet is as high as 1.82 eV which is closely coincides with the experimental value reported by Li et al.19)
Sometimes the presence of one kind of molecule affects the adsorption of a molecule of another kind,54-55) as well as negatively charged NPs could lead to strong binding of O2 molecules.49-51) Under operating conditions of CO oxidation, the catalysts are exposed to an O2-CO mixture. A CO molecule plays a role of an electron donor, and an O2 molecule does a role of an electron acceptor;49) thus, co-adsorption of CO and O2 molecules or sequential adsorption of CO and O2 may enhance the O2 adsorption chemistry on Au NPs. Table 2 shows adsorption energies of CO on Au38-TOh, Au55-Ih, and Au55-COh. These NPs strongly bind CO molecules. According to Mulliken charge analysis on the Au38-TOh, a CO molecule indeed donates electrons to Au NP (Fig. 4). These electrons are distributed to nearby Au atoms. Specifically, about 46 % of electrons are localized to the Au atom that directly interacts with the CO molecule (Au1 in Fig. 4), and 16 % of electrons are localized to the sub-surface Au atom (Au2 in Fig. 4). In Table 2, we also found that an electron donation from a CO molecule to unsupported Au NP is insufficient to facilitate O2 adsorption. Variation in the energy of O2 adsorption in the presence of a pre-existing CO molecule is marginal (see Table 2). We found that the CO-O2 co-adsorption is not a main factor that originates the excellent CO oxidation activity of Au NPs.
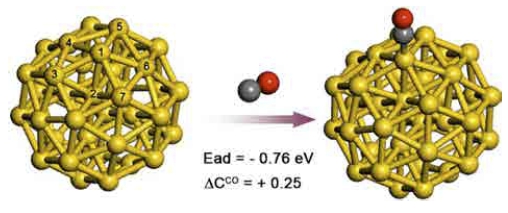
Fig. 4
Electron donation by CO adsorption. A CO molecule donates 0.25 excess electrons to the Au38-TOh (ΔCCO). Most of the excess electrons are distributed to seven numbered Au atoms. Of these, two Au atoms, Au1 and Au2, draw about 46 % and 16 % of electrons, respectively. Here color codes are gold = Au, gray = carbon, and red = oxygen.
We found that the exceptional catalytic activity of supported Au NPs does not mainly originate from the intrinsic nature of crystalline Au NPs larger than Au13. We think that the contributions of the following factors are presumably essential for the exceptional catalytic activity of Au NP catalysts.
(1) The NP-support electronic interaction. The Au NP is preferentially located on the defects of supporting oxides.18-19,22,56-57) Therefore, an electron interaction between an Au NP and the supporting material is inevitable. Moreover, in-depth studies on such electronic interaction converge to the question of how the supporting material charges Au NPs and what type of charged Au species is the reactive center for CO oxidation. The answer is, however, still unclear. We speculate that the oxygen vacancies on the supporting oxide can donate some charge to Au atoms at the NP-support interface and that these negatively charged Au atoms may enhance O2 binding.
(2) The NP-support interface as a binding site for an O2 molecule. Previous computational studies have already reported that such O2 adsorption at the NP-support interface is feasible.19,22) The NP-support interface may contribute to O2 adsorption by providing a new O2 adsorption site. In this case, a metal ion of the supporting oxide or an oxygen vacancy near the NP-support interface would anchor a gas-phase O2 molecule.
(3) Structural fluxionality of Au NPs. This is limited to very small Au NPs.19,22) Our Au13-Ih is a typical example of how the structural fluxionality of Au NPs affects their oxygen adsorption chemistry (Fig. 3).
Note that, for our unsupported Au NPs, the first two factors produced by the NP-support interface are completely excluded. We found that the unsupported Au NPs larger than the Au13-Ih (involving the Au13-COh) cannot bind O2 sufficiently strongly to ensure CO oxidation activity. Therefore, if one finds that somewhat large supported Au NPs catalyze CO oxidation, the first two factors are responsible for their CO oxidation activity. The activity does not come from Au NPs themselves. On the other hand, the Au NPs that are sufficiently small for the adsorption-driven structural change to occur are good catalysts themselves, irrespective of the contributions of supporting materials. The computational finding of Remediakis et al.22) that the rutile-TiO2-supported Au10 NP can bind O2 molecule without participation of the NPsupport interface supports our statement that such very small Au NPs can catalyze CO oxidation without the help of supporting materials.
4. Conclusion
In summary, as functions of the size, coordination number, and shape of unsupported Au NPs, we studied the oxygen adsorption chemistry of various unsupported Au NPs around 1 nm. We found that none of these factors can dominantly control the catalytic activity of small Au NPs. There is no clear quantitative relationship between the energy of O2 adsorption and the coordination number, size, or morphology Au NPs composed of 13 to 55 atoms (the Au13-Ih is an exception). We found that energetically most favorable O2 adsorption is observed in the Au13-Ih, the smallest Au NP, with the help of the structural fluxionality of such very small Au NPs. From the recent experimental studies reporting that tiny Au NPs composed of around 10 atoms are the reactive species for CO oxidation, our computational study indicates that such experimental results are originated from the structural fluxionality of tiny Au NPs. We also suggest that there is the size-threshold in Au NPs, where the smaller Au NPs can catalyze CO oxidation without the help of supporting materials whereas the support-NP interaction is required to activate larger Au NPs.
