

1. 서 론
자동차 및 산업기계 부품에서 적용되는 진공침탄 열처리는 일반적으로 900~1,050 °C 온도까지 가열한 후 아세틸렌가스를 펄스 방식으로 주입하여 강재 표면에 탄소를 확산시킨 후 퀜칭 공정을 통해 제품 표면을 경화시키는 공정이다. 기존 상압침탄과 다르게 철을 촉매로 비평형 상태에서 빠른 침탄 반응이 일어나기 때문에 비교적 낮은 압력과 아세틸렌 유량이 요구되며 높은 처리 온도에 의한 공정 시간 단축 등의 장점을 가지고 있다. 하지만 분위기 CP (carbon potential)제어가 어렵고 실시간으로 carbon flux를 측정하기 어렵기 때문에 침탄 변수에 따른 침탄 제품 내에 고용된 탄소 농도를 기준으로 공정 모델링이 이루어진다.1) 국내에서는 침탄 제품의 탄소 정량분석 기술이 확립되지 않았기 때문에 침탄 산업에서는 간접적으로 nital 부식액을 이용하여 침탄 제품 단면부를 에칭시켜 침탄 깊이를 관찰하거나 550 Hv를 가지는 특정 지점을 유효 경화깊이로 판단하여 침탄 정도를 확인하고 있다.2) 유효 경화깊이란 침탄에 의해 철 내에 침투된 탄소의 고용 효과뿐만 아니라 모재가 구성하고 있는 합금원소에 의한 경화능과 관련하여 냉각 공정을 거쳐 결정되기 때문에 철에 고용된 탄소농도를 대변하기 힘들다고 판단된다. 반면에 침탄으로부터 고용된 탄소는 고용체 강화, 석출강화, 상변태 강화 등 여러 강화기구에 영향을 미칠 뿐만 아니라 탄소가 확산된 실제 깊이를 의미하기 때문에 침탄 제품에서 요구되어지는 기계적 물성을 결정하는 중요한 인자이다.3) 따라서 다양한 침탄 조건에서 제품 내에 탄소농도에 따른 침탄 물성의 효과를 면밀히 해석하기 위해서는 신뢰성있고 정확하게 탄소농도를 분석하는 연구가 필요하다.
부품 종류(자동차 부품, 기계구조 부품, 중장비 부품 등)에 따라 요구되는 침탄 깊이의 범위는 0.3~2.0 mm를 가지며, 이와 같이 다양한 침탄깊이를 가지는 침탄 제품의 효율적인 탄소농도 분석이 가능하기 위해서 성분분석 장비는 다음과 같은 3가지 특성이 만족해야 한다. 미량의 탄소농도를 정량적으로 분석하기 위한 ‘정확성’, 제품의 다양한 위치에 탄소농도를 편리하게 연속적으로 분석하기 위한 ‘선택성’, 현실적인 범위에서 분석에 소비되는 시간 및 비용이 적절하기 위한 ‘경제성’이 충족되어야 한다. Table 1은 탄소 성분 분석을 목적으로 하는 분석 장비들의 원리, 특징 및 단점을 나타내고 있다. 시편 내의 탄소농도 검출을 위한 대표적인 분석기법으로는 carbon-sulfur 분석(CS), 글로우 방전 질량 분석(GDMS), 에너지 분산분광법(EDS), 전자빔 미세분석(EPMA)가 있다. CS 분석은 0.6 ppm의 정교한 검출한계를 가지지만 bulk 시편을 녹여 탄소농도를 분석하기 때문에 ‘선택성’에 한계가 있다.4) GDMS는 검출한계가 1 ppm으로 정확한 성분 분석이 가능하지만 0.08 µm/s의 느린 식각속도를 가져 많은 분석 시간 및 비용이 발생하기 때문에 ‘경제성’이 결여된다.5) EDS는 빠른 분석 시간과 위치별 프로파일 분석이 가능하다는 장점이 있지만 높은 검출한계(100 ppm)를 가지기 때문에 ‘정확성’에 한계가 있다.6) EPMA는 검출한계가 10 ppm으로 미량 원소의 분석이 가능하지만 시편 준비가 까다로우며 적절한 표준시편 선정과 분석조건 최적화가 요구되어진다.7,8)
Table 1.
Specifications of carbon concentration analysis apparatus.
다양한 탄소 분석장비를 사용하여 침탄강의 탄소농도 분석에 관련된 여러 연구들이 진행되어 왔다. Vontorová and Váňová9)는 Mn-Cr 저합금강을 가스침탄을 통해 약 0.8 mm 경화깊이를 가지는 침탄강의 단면 깊이에 따른 탄소농도 구배를 GD-OES 장비를 사용하여 분석하였으며 경도 및 미세조직간의 상관 관계를 규명하였다. 하지만 탄소농도 분석을 위해 약 5시간의 긴 분석 시간이 요구되었고 유효경화깊이에 상응하는 탄소농도를 도출하기에 분석 횟수가 부족하였다. Robaut et al.7)은 EPMA 장비를 사용하여 X-ray intensities의 k ratio를 분석하여 φ(ρz) model을 적용한 기존 탄소 정량분석에 대한 한계를 규명하였고 표준 시편에 대한 중요성을 강조하였다. Peng et al.10)은 SUS 316L 강을 저온 침탄시켜 표면에 고용되어있는 탄소농도를 EPMA 장비를 통해 분석하였으며 잔류응력에 따른 탄소 확산의 영향을 연구하였다. Kula et al.11)은 C20 저합금강을 진공침탄 공정을 거쳐 CS 분석을 통해 탄소 포화 농도 및 단면깊이에 따른 탄소농도 구배를 분석하였다. CS 분석은 정확한 탄소 정량 분석에 있어서 탁월한 분석방법이라고 판단하였지만 침탄강을 일정 간격으로 얇은 chip을 내어 탄소농도 구배를 분석하기에 비교적 효율적이지 못하다고 생각된다.
이에 본 연구에서 침탄 제품의 탄소농도 프로파일 분석을 위해 EPMA를 사용하여 기존 분석방법에 따른 정량 분석 한계를 규명할 뿐만 아니라 보완된 분석 모델링을 적용하여 침탄 제품의 탄소농도를 보다 정확하고 신뢰성 있는 분석 결과를 도출하였다.
2. 실험 방법
본 연구에서 탄소농도 분석에 대상인 침탄 시편을 제작하기 위해 사용된 시험편은 지름 30 mm, 높이 10 mm의 디스크 형상을 가지는 AISI 4115 강이다. 시험편이 구성하는 화학 성분은 Fe-0.163C-0.924Cr-0.172Mo-0.234Si-0.624Mn-0.121Ni-0.126Cu (wt%)이다. 침탄 공정 과정으로는 수평식 석영로 안에 시험편을 장입하고 0.01 torr까지 진공 배기하면서 950 °C까지 승온하여 30분 동안 등온 유지한 후, 로 내 압력을 10 torr로 제어하면서 아세틸렌가스(C2H2, 99.99 %)를 6분 동안 20 sccm 유량으로 주입한 후, 80 °C에서 오일에 30분 동안 급냉하였다.
탄소농도 분석장비 종류에 따른 침탄 시편의 탄소농도 결과를 비교하기 위해 CS (ELTRA, CS-800), GDMS (MSI, GD90RF), EDS (THERMO SCI, EM-30AX) 및 EPMA (JEOL, JXA-8500F)를 사용하였다. CS 장비는 시편을 조연제와 함께 도가니에 넣고 가열시켜 형성된 CO, CO2에 의한 적외선 흡수량 변화를 통해 탄소 함량을 정량적으로 검출한다. 각 부위별 탄소농도를 분석하기 위해 침탄 시편의 표면으로부터 200 µm 간격마다 chip 형태로 가공하였으며 인가 전압, 가열 온도 및 산소 가압 조건은 각각 230 V, 2,000 °C, 4 bar로 설정하였다. GDMS 장비는 글로우 방전을 발생시켜 시편 표면을 단위시간에 따른 깊이만큼 식각하여 이온화된 성분을 질량 분석기로 측정한다. 침탄시편의 표면을 0.08 µm/s 속도로 식각하여 연속적인 탄소농도 프로파일을 분석하였으며 RF 방식으로 인가전압 및 Ar 압력을 각각 700 V, 100 Pa 조건으로 설정하였다. EDS 장비는 입사전자가 시료 표면에 충돌하면서 발생되는 특성 X선을 검출하여 성분 함량을 분석한다. 분석조건은 가속전압, 체류시간 및 working distance를 각각 15 kV, 18 ms, 15 mm으로 설정하여 50 µm 간격마다 전자빔을 조사시켜 탄소농도 프로파일을 분석하였다. EPMA 장비는 가속된 전자빔을 시료를 구성하고 있는 원자와 충돌시켜 시료의 구성원소들로부터 고유한 파장을 갖는 X선을 검출하여 화학조성을 규명한다. 진공도, 가속전압 및 빔 전류를 각각 10-7 torr, 10 keV, 50 nA로 설정하였으며 침탄 시편 표면으로부터 50 µm 간격으로 전자빔을 조사하였다. 표준시편으로 사용된 순철(0.008 wt%C)과 그라파이트(99.98 wt%C)에 의해 도출된 calibration curve를 사용하여 침탄 시편으로부터 측정된 Kα intensity를 탄소농도로 산출하였다.
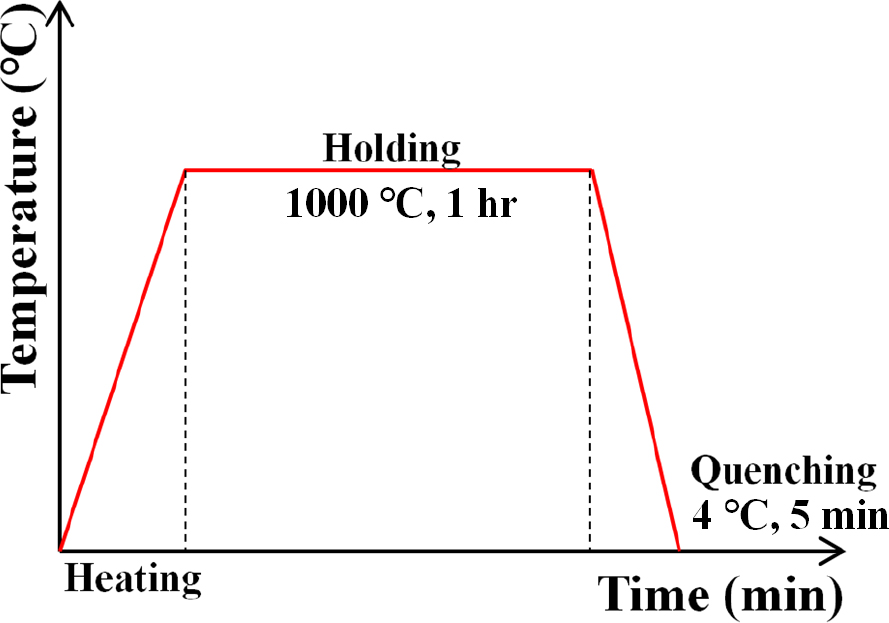
EPMA를 사용한 기존의 탄소농도 정량 분석의 정확성과 신뢰성을 높이기 위해 탄소농도별 AISI 8620 (0.2 wt%), AISI 4140 (0.4 wt%), AISI 1065 (0.6 wt%), AISI 52100 (0.98 wt%) 강들을 표준시편으로 선정하였다. 본 표준시편은 지름 30 mm, 높이 10 mm의 디스크 형상을 가지며 조성은 Table 2와 같다. 표준시편의 탄소 균질화 처리를 위해 각 시편들을 Fig. 1과 같이 1,000 °C에 가열 및 1시간 등온 유지시킨 후에 4 °C의 물에 5분 동안 급냉하였다. 또한 개선된 calibration curve를 도출하기 위해 가속전압(10, 15, 30 keV), 빔 전류(20, 50 nA)로 설정하여 분석조건에 따른 표준시편의 Kα intensity 변화를 관찰하였다. 개선된 calibration curve를 적용하여 unknown sample로서 침탄 시편의 단면깊이별 탄소농도 프로파일을 분석하였고 기존 calibration curve로부터 측정된 탄소농도 프로파일과 비교하였다.
Table 2.
Chemical compositions of standard samples (wt%).
3. 결과 및 고찰
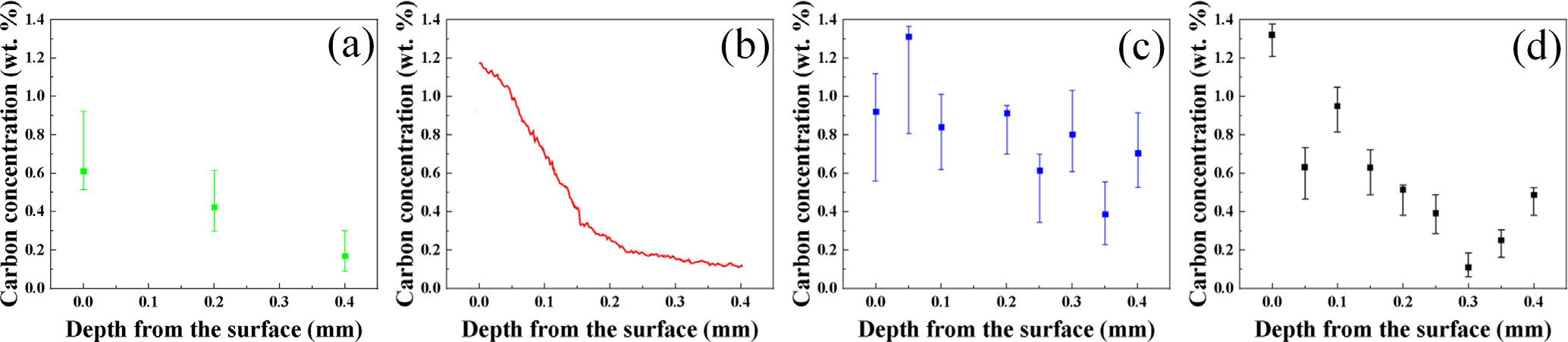
하지만 앞서 언급한 각각의 검출기법의 장단점이 존재하여 분석 데이터의 정확성을 향상시키기 위한 분석기법 선정을 위해 Fig. 2와 같이 탄소 성분분석 장비 종류에 따른 침탄시편의 탄소농도 프로파일을 비교 분석하였다. CS 장비로부터 분석된 탄소농도는 침탄 제품이 가지는 탄소 고용범위 내에 있기 때문에 신뢰성 있는 결과라고 판단하지만 분석하고자 하는 특정 부위의 chip 가공작업이 필요하며 정확한 부위의 탄소농도를 분석하기 어렵다[Fig. 2(a)]. GDMS 분석의 경우, 시험편의 전처리가 필요없이 단위 시간에 따라 미세한 두께로 시편 표면을 식각하기 때문에 위치별 연속적인 탄소농도 프로파일을 얻을 수 있었으며 분석의 정확도는 우수하였다[Fig. 2(b)]. 반면에 EDS 및 EPMA 분석은 전자빔을 원하는 위치에 쉽게 조사하여 탄소성분을 빠르게 측정할 수 있는 반면에 신뢰할 수 없는 탄소농도 결과를 얻었다. EDS 분석의 경우, 피크 중첩이 심할 뿐만 아니라 P/B (peak to background)가 낮기 때문에 침탄 제품에 미량의 탄소농도를 분석하기에 적합한 분석 방법이 아니라고 판단된다[Fig. 2(c)]. EPMA 정량 분석은 WDS 방식을 사용하여 EDS보다 비교적 정확하지만 calibration curve에 사용된 표준시편의 탄소농도가 철의 탄소 고용범위 내에 미치지 않기 때문에 분석 정확도가 낮다고 판단된다[Fig. 2(d)].
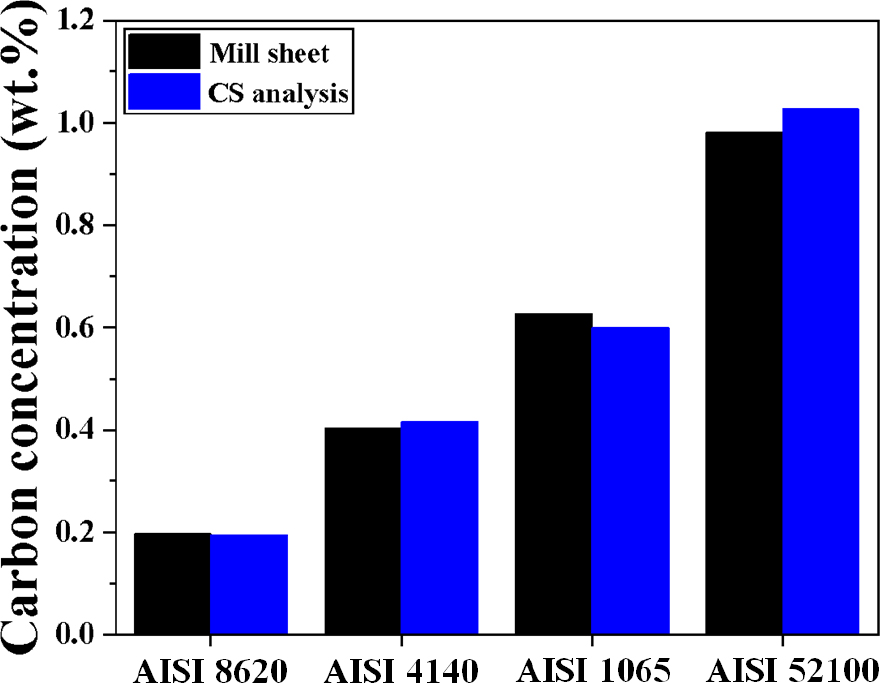
EPMA 분석에서 미량의 성분 분석과 관련된 ‘정확성’을 보완하기 위해 기존 calibration curve에 사용된 pure iron 및 graphite 대신에 침탄 제품이 가지는 탄소농도 범위를 기준으로 선정된 AISI 8620, AISI 4140, AISI 1065, AISI 52100 강을 표준시편으로 사용하였다. Fig. 3은 탄소농도 별로 선정된 4가지 표준시편의 Mill sheet에 명시된 탄소농도 및 CS분석을 통한 탄소농도를 보여주고 있다. CS 분석 결과로 AISI 8620, AISI 4140, AISI 1065, AISI 52100 강의 탄소농도는 각각 0.193, 0.415, 0.599, 1.027 wt%를 가지며 Mill sheet의 명시된 탄소농도와 비슷한 결과를 나타내고 있다. 본 연구에서 calibration curve로 사용되는 표준시편의 탄소농도는 CS 분석으로부터 분석된 결과를 사용하였다.
탄소농도에 따른 Kα intensity가 다르기 때문에 신뢰성 있는 calibration curve 도출을 위해서는 표준시편의 위치에 따른 탄소농도가 균일해야 한다. Fig. 4는 표준시편을 균질화 열처리를 진행하기 전후의 미세조직 사진을 나타내고 있다. 균질화 열처리 전의 미세조직의 경우, 저합금강인 AISI 8620, AISI 4140, AISI 1065강은 페라이트 및 펄라이트로 구성된 혼합조직이 관찰되었고 공석 조성 내의 표준시편의 탄소농도가 높을수록 시멘타이트 분율이 높아지기 때문에 초석 페라이트 분율이 적어지고 펄라이트 분율이 높아진다. 특히, 베어링강인 SUJ2강은 페라이트 및 구형 탄화물이 관찰되었다. 반면에 균질화 처리 후의 미세조직의 경우, 모든 표준시편에서 대부분 탄소가 균일한 마르텐사이트가 관찰되었다.

Fig. 5는 EPMA 장비에서 가속전압 및 빔 전류를 각각 15 keV, 20 nA로 설정하고 균질화 열처리 유무에 따른 표준시편의 단면부를 100 µm 간격으로 전자빔을 조사하여 검출한 Kα intensity 값이다. 균질화 처리를 하지 않은 표준시편의 경우, 함유하고 있는 탄소농도가 높은 강종일수록 전반적인 Kα intensity는 높아진다. 강종에 상관없이 대부분 위치의 Kα intensity값은 불균일한 결과를 얻었는데 이는 미세조직 및 탄소가 위치별로 불균일하게 분포하기 때문이다. 반면에 균질화 처리된 표준시편의 경우, 위치에 따라 탄소가 균일하게 분포하기 때문에 Kα intensity값은 수렴에 가깝게 측정되었다. 표준시편의 탄소농도가 높아질수록 위치별 Kα intensity의 균일도는 높아짐을 확인할 수 있는데 이는 탄소농도가 높을수록 경화능에 의해 마르텐사이트 조직이 형성되기 쉽기 때문에 위치에 따른 탄소농도 균일도가 높아지는데 기인한다.
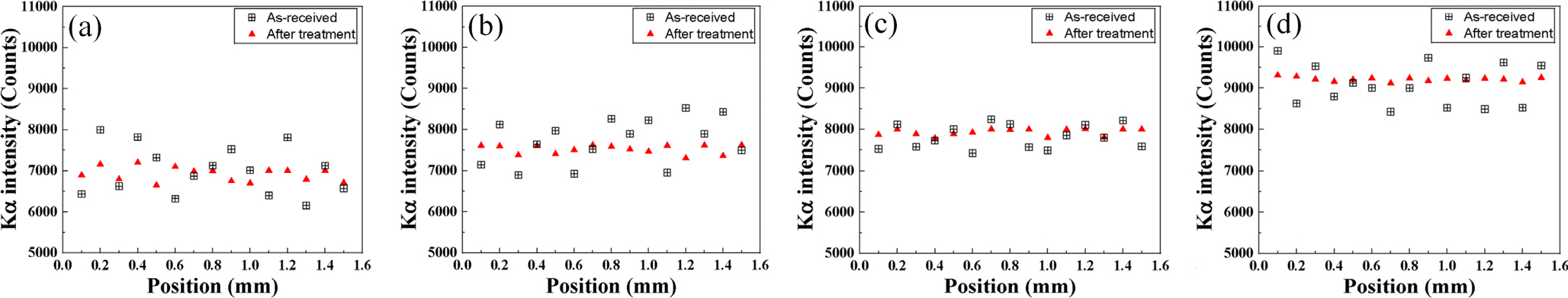
Fig. 6은 빔 전류를 20 nA로 설정하였고 가속 전압 조건(10, 15, 30 keV) 및 전자빔의 조사 시간에 따른 균질화 처리된 표준시편의 Kα intensity를 보여주고 있다. 10 keV 조건의 경우, 조사 시간에 따라 Kα intensity이 모든 강종에서 2~5 % 범위의 오차를 가지며 변화하고 있다. 이는 낮은 가속전압을 가진 전자빔이 조사되어 시편 표면에 작은 interaction volume 및 낮은 입사에너지가 가해져 불안정한 X선 검출이 일어난다. 또한 30 keV 조건의 경우, 높은 가속전압에 의해 높은 입사에너지와 큰 interaction volume에 의해 연속 X-선 및 후방 산란전자와 같은 부산물에 의해 intensity가 높지만 불안정한 X선이 검출되었다. 따라서 오차를 갖는 Kα intensity로부터 설정된 calibration curve은 탄소 정량분석에 사용하기에는 부적절하다 판단된다. 반면에 15 keV 조건의 경우, 적절한 입사에너지와 interaction volume으로부터 비교적 Kα intensity가 균일하면서 많이 검출되었다.
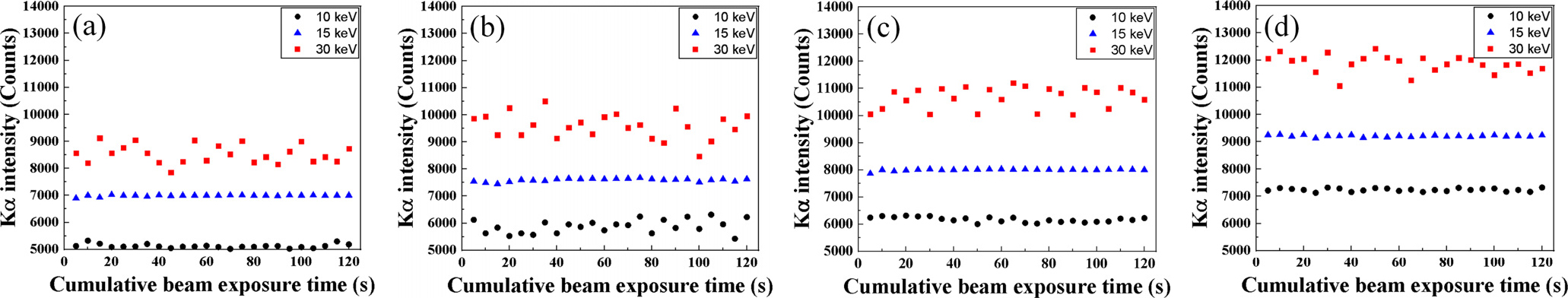
Fig. 7은 Monte Carlo simulation software를 사용하여 계산된 가속전압에 따른 강재 표면부의 interaction volume을 나타내고 있다. 10 keV, 15 keV, 30 keV 조건에서 전자빔이 영향을 주는 penetration depth는 각각 2.3, 3.1, 4.8 µm로 계산되었다. 이는 가속전압이 높아짐에 따라 전자빔 에너지 및 전자 분산이 커지기 때문에 시료내로 더 깊숙이 침투하여 interaction volume은 커진다. 가속전압을 높일수록 넓은 면적에 대한 평균적인 Kα intensity를 측정할 수 있는 장점이 있음에도 불구하고 불필요한 두께까지 측정되거나 전자빔에 의한 시편 손상이 있기 때문에 분석하고자 하는 시편의 밀도 및 미세조직을 고려하여 적절한 interaction volume을 가지기 위한 최적의 가속전압이 설정되어야 한다.
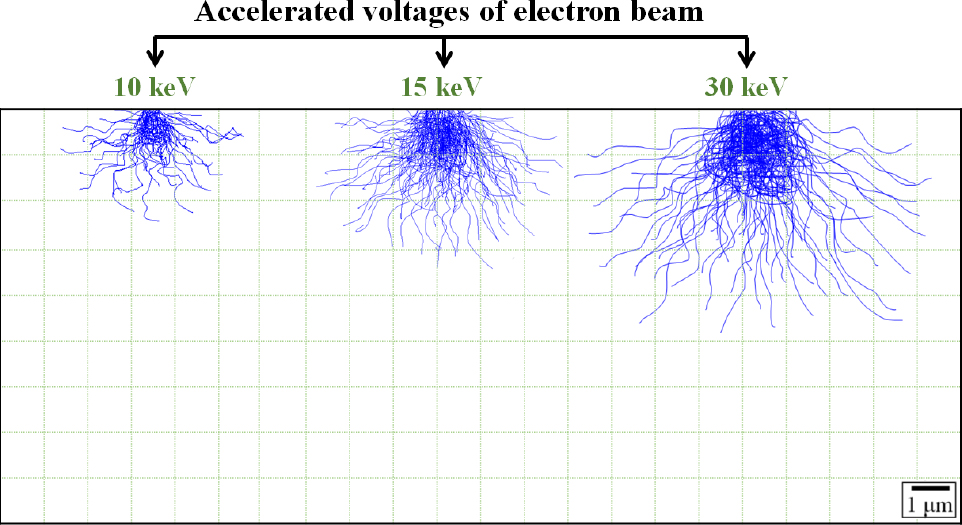
Fig. 8은 가속전압은 15 keV로 설정하였고 빔 전류(20, 50 nA) 및 전자빔의 조사시간에 따른 균질화 처리된 표준시편의 Kα intensity 변화를 보여주고 있다. 빔 전류 20 nA의 경우, 모든 표준시편에서 Kα intensity은 변동성 없이 수렴하는 경향을 보였고 함유하는 탄소농도가 높은 표준시편일수록 Kα intensity는 비례하여 높아진다. 반면에 빔 전류 50 nA를 가진 조건에서 조사시간에 따른 Kα intensity는 ±5 % 정도 오차 변동을 가진다. 이는 높은 전류밀도를 가지는 전자빔으로부터 시편의 손상에 의해 intensity fluctuation이 발생된다고 판단된다. 따라서 가속전압 및 빔 전류에 따른 안정적이고 적절한 intensity가 검출되는 EPMA 분석 최적조건은 15 keV (가속전압), 20 nA (빔 전류)로 선정하였다.
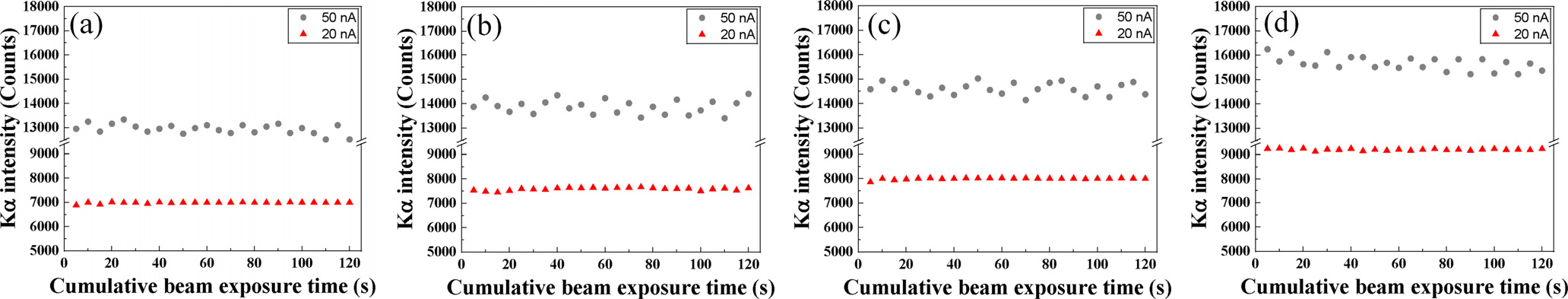
Fig. 9는 표준시편으로서 pure iron과 graphite [Fig. 9(a)] 및 본 새로 선정된 탄소농도 별 표준시편을 사용한 calibration curve [Fig. 9(b)]를 보여주고 있다. 기존 calibration curve의 경우, pure iron 및 graphite 표준시편에서 각각 Kα intensity가 4952.24, 42874.11 counts로 측정되었으며 기울기 및 y 절편이 각각 4237.9187, 4952.2433를 가지는 calibration curve가 도출되었다. 보완된 calibration curve의 경우, 각 탄소농도(0.193, 0.415, 0.599, 1.027 wt%)에 따른 평균 Kα intensity는 각각 7026.55, 7566.37, 7969.03, 9225.66 counts로 측정되었으며 기울기 및 y 절편이 각각 2656.2761, 6538.1545를 가지는 calibration curve가 도출되었다. 보완된 calibration curve은 침탄 제품에 탄소가 실제 고용되어있는 범위를 가지고 있기 때문에 기존 조건보다 비교적 신뢰성이 높은 탄소농도 분석이 가능할 것으로 판단된다.
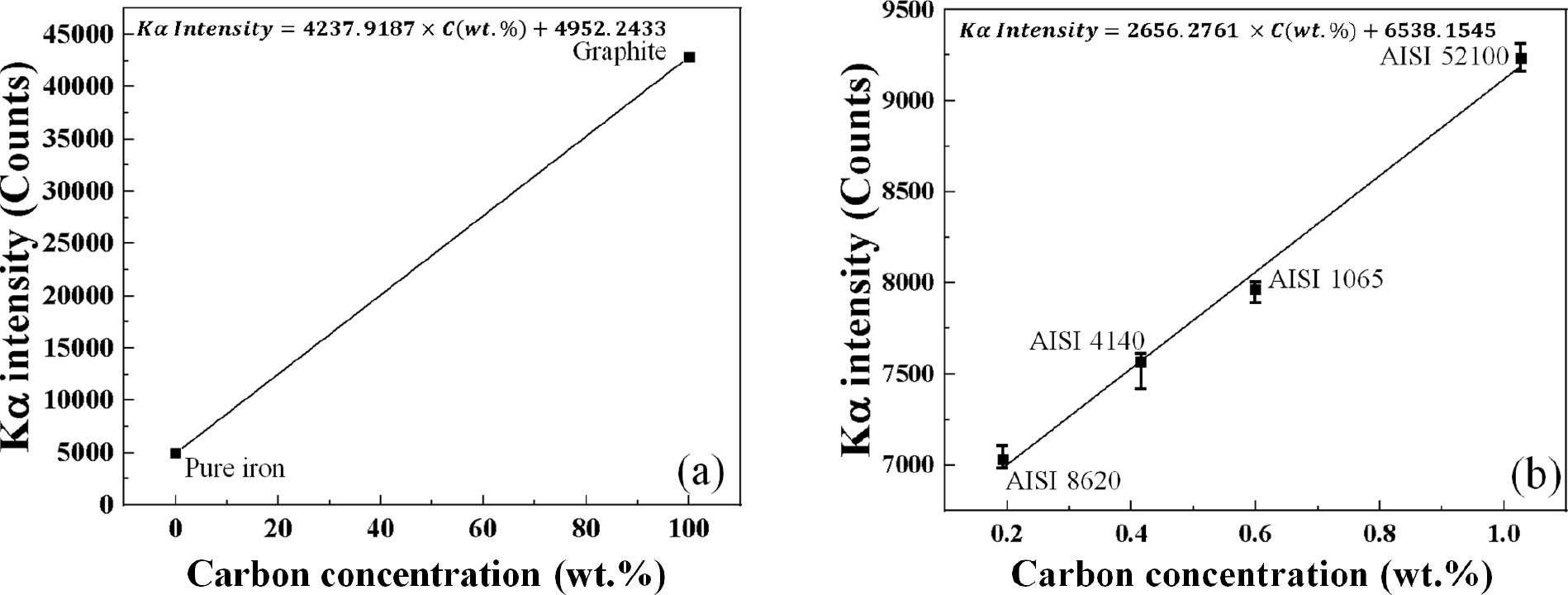
Fig. 10(a)와 같이 950 °C 온도에 6분 침탄 처리된 시편의 단면에서 표면으로부터 약 150 µm 부위에 EPMA 전자빔을 조사하여 검출된 intensity를 기존 calibration curve와 보완된 calibration curve를 통해 탄소농도를 산출하였다. 해당 부위에 Kα intensity는 7663.08 counts로 검출되었고 calibration curve로부터 산출된 탄소농도는 0.632 wt% (기존), 0.421 wt% (보완)이다. 보완된 curve로부터 탄소농도는 Fig. 2(a)의 GDMS로부터 측정된 150 µm 부위의 탄소농도 데이터와 비교적 비슷하고 reasonable한 결과로 판단되는 반면에 조사된 부위의 미세조직을 보더라도 기존 curve로부터 산출된 탄소농도는 타당하지 않다. Fig. 10(b)는 침탄시편 단면 미세조직에서 전자빔이 조사된 부위는 조사 직경을 1 µm로 설정하였음에도 불구하고 시편과 가속 전자의 접촉으로 약 16 µm의 degradation이 일어난다. Degradation 정도는 전자빔과 관련하여 가속전압, 빔 전류에 따라 상이하며, 조사되는 시료의 밀도에 따라 결정된다. 전자빔에 의해 형성된 degradation 부위는 표면 변질에 의해 Kα intensity가 변할 수 있기 때문에 분석 정확도가 떨어질 수 있다. 따라서 분석 범위가 degradation 영역을 간섭받지 않을 만큼 간격으로 탄소농도 분석이 이루어져야 한다.
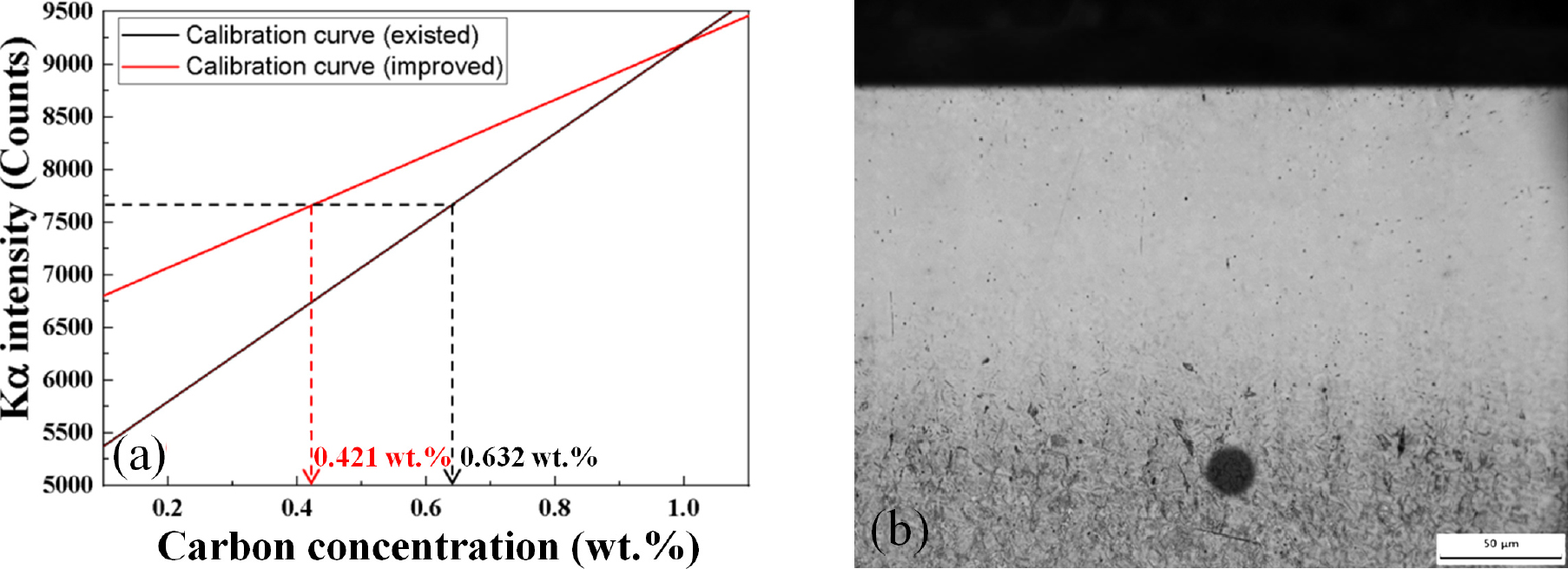
Fig. 11과 같이 950 °C 온도에 6분 침탄 처리된 AISI 4115 시편의 단면에서 표면으로부터 0.02 mm 지점을 시작으로 0.05 mm 간격으로 전자빔을 조사하여 측정된 Kα intensity를 기존 및 보완된 curve에 의해 위치별 탄소농도 프로파일을 산출 하였다. 침탄시편 표면으로부터 0.02 mm 지점의 최대 탄소농도는 1.39 wt% (기존), 1.08 wt% (보완)로 측정되었으며 탄소농도가 일정한 모재부위에서 기존 curve로부터 산출된 탄소농도는 0.08~0.5 wt%로 신뢰할 수 없는 결과이다. 특히, 침탄에 의해 탄소가 고용된 0~0.2 mm 구간에서 GDMS 장비로 측정한 탄소농도와 비교할 때 기존 curve로부터 산출된 탄소농도의 오차는 전반적으로 27~30 %이다. 반면에 보완된 curve로부터 산출된 탄소농도는 모든 위치에서 평균적으로 4~6 %의 오차 범위를 가진다. 기존 EPMA 분석법으로부터 탄소농도 정량 분석을 보완하기 위해 침탄 제품의 탄소농도 범위 내에 표준시편을 선정하고 적은 fluctuation와 검출감도가 높은 intensity를 갖는 최적 분석조건을 도출하여 보다 정확하고 신뢰성 있는 탄소농도 결과를 확인하였다.
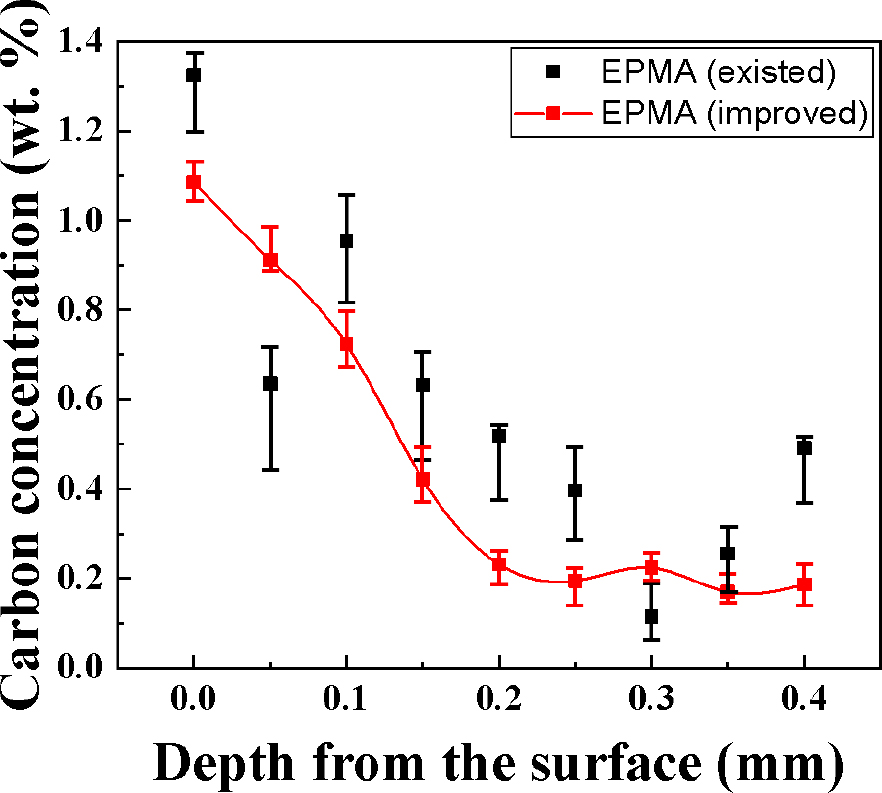
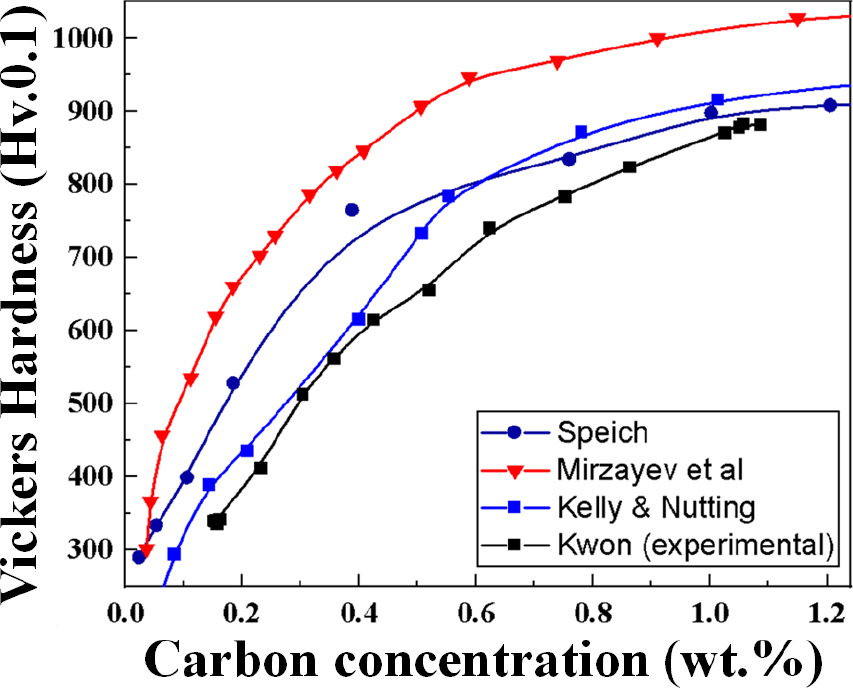
Fig. 12는 탄소농도에 따른 저합금강을 퀜칭시킨 후 측정된 경도의 효과에 관한 연구로 Speich and Warlimont,12) Mirzayev et al.,13) Kelly and Nutting14)은 1960년부터 다양한 실험을 통해 model 식을 도출하였다. 본 실험에서 측정한 침탄시편의 탄소농도에 따른 비커스 경도와 문헌의 model들을 중첩시켜 비교하였다. 실험을 통한 탄소농도에 따른 경도 경향은 수냉 냉각 조건이 Kelly and Nutting model과 가장 유사하며 이는 다른 model들에 비해 오스테나이징 온도(950 °C) 및 냉각 조건이 비슷하기 때문이다. 실험에 의한 탄소농도 별 경도 구배는 전반적인 model들과 비슷하게 탄소농도가 높아질수록 경도는 증가한다. 특히 탄소농도가 0.2~0.6 wt%를 가지는 구역은 monotonical하게 경도가 증가하는데 이는 sub-grain 미세화와 C-rich phase의 증가에 의한 primary martensite 변태에 기인한다. 또한 탄소농도 0.9~1.0 wt% 이상에서는 전반적으로 다른 경향을 보이며 기존 BCT martensite에서 retained FCC austenite 분율이 늘어나기 때문에 경도 향상의 효과가 감소한다.
4. 결 론
철 내 오스테나이트 상의 탄소 고용 범위에 준하는 표준시편을 선정하여 가속전압 및 빔 전류 조건에 따라 측정되는 Kα intensity 변화를 살펴보고 calibration을 보완하여 침탄 시편의 단면깊이 탄소농도 프로파일을 측정하였고, 주요 결론은 아래와 같다.
(1) 1 wt% 이내 수준의 탄소농도 분석은 GDMS 분석이 가장 정확하며, 기존 EPMA 분석에 사용된 표준시편은 pure iron (0.008 wt%), graphite (99.98 wt%)로 탄소 분석의 정확성이 저조하다.
(2) 균질화 열처리를 통해 선정된 표준시편의 위치별 Kα intensity 균일도는 85 %에서 97 %로 증가한다.
(3) EPMA 전자빔 조건으로서 가속전압을 15 keV까지 높일수록 보다 안정하고 높은 intensity가 검출되지만 30 keV 조건에서 intensity fluctuation이 일어난다. 또한 빔 전류를 높일수록 시료의 손상에 의해 불안정한 intensity를 갖는다.
(4) EPMA 분석 최적조건으로서 가속전압 및 빔 전류는 각각 15 keV, 20 nA으로 설정하였다.
(5) 보완된 calibration curve의 식은 “Intensity = 2656.2761 X C (wt%) + 6538.1545”로 도출되었으며 기존 curve로부터 산출된 탄소농도 오차율 27 %에 비해 약 4 %의 낮은 오차율로 보다 정확한 탄소농도 분석에 대한 데이터를 도출하였다.
