

1. Introduction
2. Computational Methods
3. Results and Discussion
3.1. High-throughput screening (HTS)
3.2. Li+ diffusion behaviors of LiPN2 and LiSi2N3
3.3. C and O atoms doping
3.4. Li+ conductivity of O-doped materials
4. Conclusion
1. Introduction
Conventional lithium-ion batteries, which rely on flammable liquid electrolytes, present significant safety concerns and face limitations in achieving substantial improvements in energy density.1,2) These challenges have driven growing interest in all-solid-state batteries, which are emerging as a promising next-generation solution due to their potential to offer both excellent stability and high energy density.3,4,5,6) A key component of these batteries is the solid-state electrolyte (SSE), known for its broad electrochemical stability, enabling the batteries to operate across a wide voltage range. The solid nature of SSEs provides exceptional heat resistance and greatly reduces the risk of leakage or explosion, significantly enhancing safety. Furthermore, this solid structure allows for a reduction in cell volume, with particularly stable performance at elevated temperatures. While ongoing research seeks to optimize performance at lower temperatures, the development of SSEs has become increasingly critical to advancing battery technology.
Current research into SSEs has focused mainly on four categories: oxides, polymer, sulfides, and halides electrolytes. Oxide-based SSEs have excellent chemical and electrochemical stability and structural robustness. Garnet-type Li7La3Zr2O12 (LLZO) and NASICON-type materials (such as LixM2(XO4)3 and Li1.5Al0.5Ge1.5(PO4)3, LAGP) are considered promising candidates for oxide-based SSEs in Li metal all-solid-state batteries due to their high stability with Li.7,8) Specifically, LLZO has an ionic conductivity of up to 5.09 × 10-4 S/cm at room temperature, and LAGP exhibits 2.83 × 10-4 S/cm, which is relatively high for solid materials, but still lower than desired for commercial applications.9) Also, these materials face issues like poor interfacial contact with lithium, leading to high resistance, and their complex, costly fabrication processes hinder large-scale application.
Polymer-based SSEs possess a flexible structure, which makes their manufacturing processes relatively simple and their production costs lower. Polymer-based SSEs are usually complexed with Li salts. The ionic conductivity of polyethylene oxide (PEO)-based electrolytes is strongly temperature dependent, with ionic conductivities ranging from 1 × 10-6 to 1 × 10-5 S/cm at room temperature, depending on the salt and additives used. Polyvinylidene fluoride (PVDF)-based electrolytes have an ionic conductivity of 0.23 mS/cm at 90 °C for the PVDF-HFP/LTFSI composite.10) They have low ionic conductivity at room temperature, which necessitates their use in high-temperature environments.
Sulfide-based SSEs have relatively high ionic conductivity and low interfacial resistivity, but they are highly sensitive to moisture, which can lead to the release of toxic gases. A notable development was the introduction of Li10GeP2S12 (LGPS) in 2011, which drew attention for its ionic conductivity of approximately 12 mS/cm, comparable to that of conventional liquid electrolytes, as well as its wide potential window.11) In 2016, further studies on materials with the same LGPS-type crystal structure (such as Li9.54Si1.74P1.44S11.7Cl0.3 Li9.6P3S12, and Li7P2S8I12)) revealed that Li9.54Si1.74P1.44S11.7Cl0.3 exhibited an even higher ionic conductivity of 25 mS/cm.13) However, challenges remain, including issues with interfacial stability due to side reactions, difficulties in large-scale production, and concerns about long-term stability.
Halide-type SSEs include Li-argyrodite (Li6PS5X, X = Cl, Br, and I),14) a modified argyrodite with high ionic conductivity for Ag ions. It is excellent in terms of stability with Li metal, and when the electrolyte comes into contact with Li, a passivation layer is formed at a low rate that improves stability.15) However, due to the nature of the manufacturing process, it is difficult to produce in large scale, and it is sensitive to moisture and carbon dioxide, which can form corrosive compounds. Another Halide-type SSE is Halospinels, such as Li2Sc2/3Cl4, which offers an ionic conductivity of 1.5 mS/cm and a 3D Li+ diffusion pathway for efficient ion transport. Li2Sc2/3Cl4 also shows high oxidative stability, enabling stable performance with high-voltage cathodes up to 4.6 V without protective coatings.16,17,18) However, its lower conductivity compared to sulfide electrolytes and reactivity with Li metal present challenges, and it requires careful handling due to moisture sensitivity. As such SSEs still face significant challenges in achieving both high ionic conductivity and electrochemical and chemical stability.
In this paper, we aimed to explore new SSE materials incorporating nitrogen, selected for its chemical stability and low reactivity. To identify these materials, we employed high-throughput screening (HTS) techniques and analyzed the transport properties of the selected candidates. Additionally, we investigated potential improvements by doping nitrogen-based structures with carbon and oxygen. Our results conclude with an in-depth analysis of the doped materials, highlighting the enhanced ionic conductivity observed in oxygen-doped structures. Overall, this research not only identifies novel nitrogen-based SSE candidates but also establishes a comprehensive methodological framework for discovering new battery materials. Our findings contribute to ongoing efforts to develop safer, more efficient, and cost-effective SSEs for next-generation energy storage technologies.
2. Computational Methods
First-principles calculations and ab initio molecular dynamics (AIMD) simulations were conducted using the Vienna Ab initio Simulation Package (VASP).19,20,21) The core electrons were represented using the projector augmented wave (PAW)22) method, while the exchange-correlation functional was defined by the Perdew-Burke-Ernzerhof (PBE)23) approach within the framework of the generalized gradient approximation (GGA). A plane-wave cutoff energy of 500 eV was applied, and the energy convergence criterion was set to 10-5 eV. Atomic forces were minimized to below 0.02 eV Å-1 until convergence was achieved. Statistical analyses of the AIMD simulations were performed using Pymatgen24) to understand Li-ion diffusion behavior. The AIMD simulations were carried out with a Nose-Hoover thermostat in a statistical ensemble with fixed particle number, volume, and temperature (NVT) conditions.25) The time step for the simulations was set to 1 fs, and each diffusion simulation was run for 100 ps at temperatures ranging from 800 K to 1,600 K, using a 3 × 3 × 1 k-point grid. The Bond Valence Sum (BVS) 3D mapping calculations26) were performed with the PyAbstantia program.27)
3. Results and Discussion
3.1. High-throughput screening (HTS)
To find new solid electrolytes beyond the conventional oxides, sulfides, and halides, we selected materials that use nitrogen as the anion as the next candidates. The search for new nitride solid electrolyte materials was conducted using a HTS technique, targeting a total of 154,718 materials listed in the Materials Project database.28) This process was carried out in five key stages, as shown in Fig. 1. Firstly, as we wanted to find Li-based electrolytes, we filtered the 154,718 materials to include only those containing Li. Second, we narrowed down the selection to nitrogen-containing materials, with additional considerations for stability, ease of synthesis, and practical applicability. To improve experimental reproducibility and synthesis feasibility, only materials with 3 to 4 elements were selected, excluding overly complex material structures. Third, to ensure that the materials are thermodynamically stable and have low formation energy, we filtered out materials based on their position on the convex hull, selecting only those that lie on it. Fourth, as electrolytes require very low electrical conductivity, only materials with a band gap greater than 3 eV were selected. Fifthly, we excluded alkali metals, which could potentially substitute Li, and lanthanide elements, which could introduce toxicity. Through this five-step screening process, 17 SSE candidate materials were identified, and their structures are shown in Fig. 2.
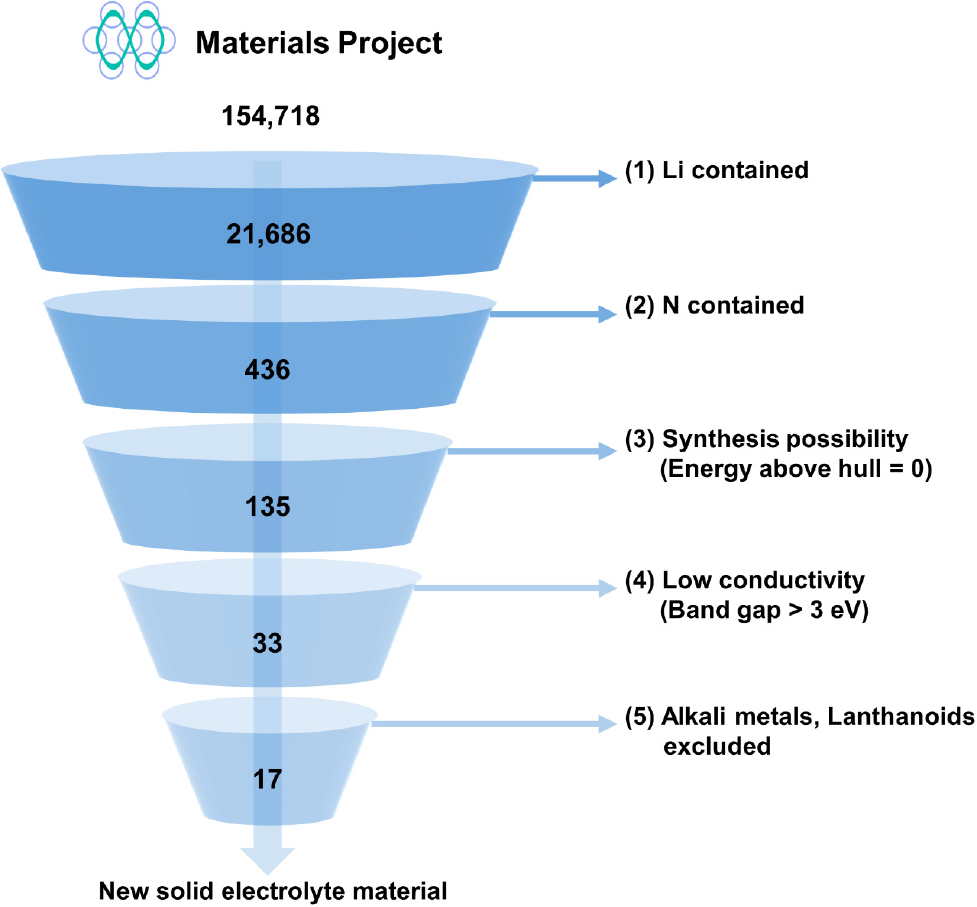
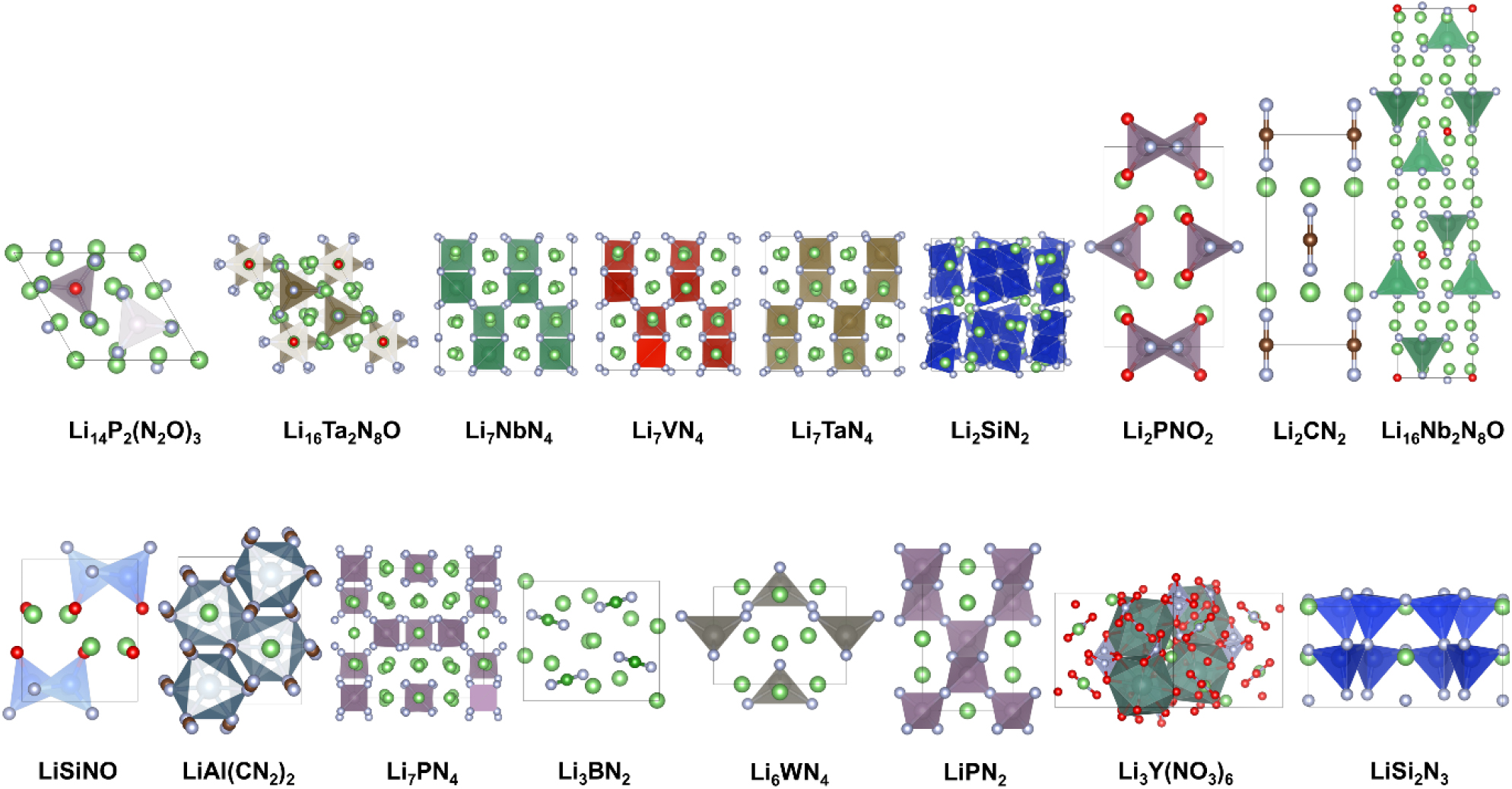
Fig. 2.
The figure above shows the 17 candidate materials selected through the HTS process in Fig. 1. These materials were filtered according to specific criteria, such as the presence of Li and nitrogen, and synthetic potential.
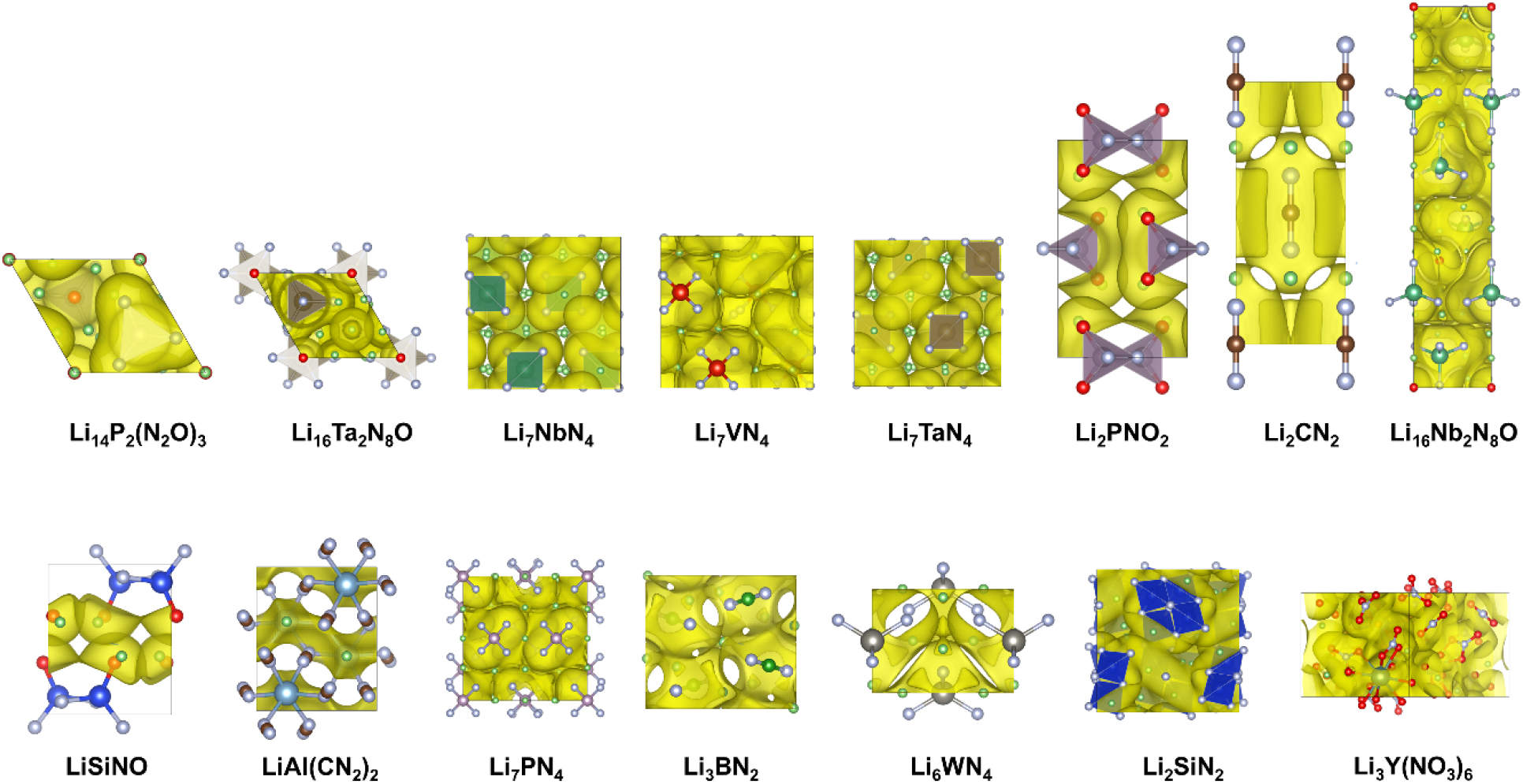
For these 17 materials, we analyzed the diffusion pathways of Li ions using the concept of BVS, which indicates the bond strength between specific atoms and surrounding atoms. The BVS values of Li ions were calculated at different points in the crystal structure, and these values were mapped in 3D to identify the most likely pathways for Li-ion movement. (Fig. 3) A more continuous and linear mapping suggests a better diffusion pathway for Li ions. Among the 17 materials, LiPN2 and LiSi2N3 showed the most linear mappings, indicating that they are more promising candidates as SSEs compared to the others.
3.2. Li+ diffusion behaviors of LiPN2 and LiSi2N3
AIMD was performed on LiPN2 and LiSi2N3, which showed favorable pathways for Li movement in the BVS, to understand the diffusion mechanism and conductivity of Li-ions. AIMD simulations were performed at elevated temperatures (800 K, 1,000 K, 1,200 K, 1,400 K, and 1,600 K), and diffusion simulations were performed at 100 ps. Since the computational cost of AIMD calculations is too high to directly obtain Li conductivity at room temperature, we calculated the room temperature Li ionic conductivity using the values obtained at high temperatures through the Arrhenius equation. Based on the AIMD results, the mean square displacement (MSD) was obtained, which is defined as follows.
where, N is the total number of Li ions in the system and represents the displacement of the i-th Li ion at time t. MSD is related to the diffusion coefficient DLi,
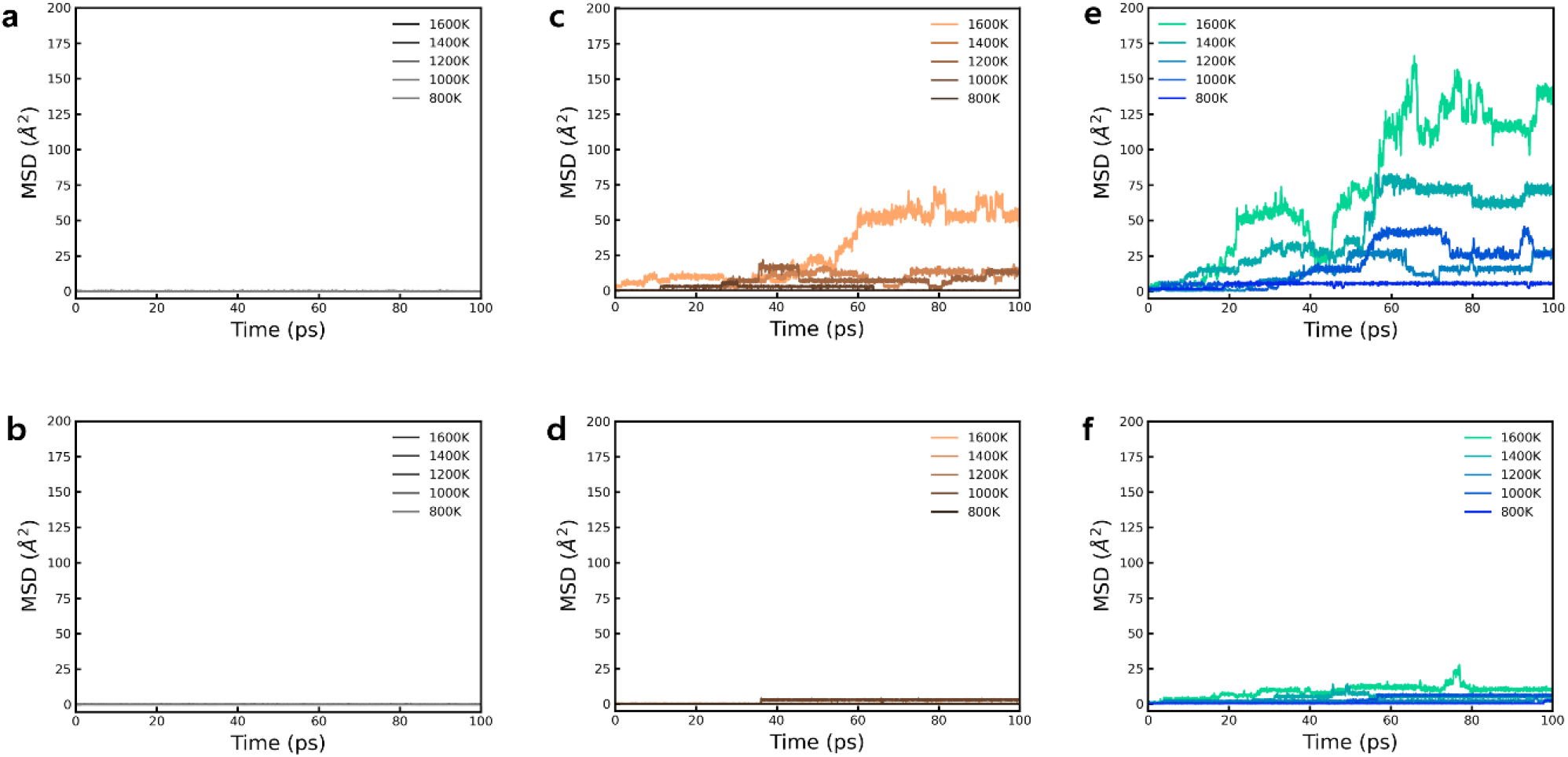
where, d represents the dimensionality of the lattice in which diffusion occurs. From the MSD analysis, it was observed that the movement of Li ions in these two materials was minimal [Fig. 4(a, b)]. In other words, these materials alone are not sufficient to function effectively as SSEs. Although the structure has a framework that allows for fast Li movement, the insufficient amount of lithium for high ionic conductivity may be a primary reason for the low ionic conductivity.29,30,31)
3.3. C and O atoms doping
To enhance the mobility of the Li ions, additional experiments were designed which one nitrogen atom in each of the two materials was doped with carbon and oxygen. It is well known that Li conductivity can be improved by controlling Li concentration through aliovalent doping, as demonstrated by materials like Li1.5Al0.5Ge1.5(PO4)3. Analogous to the doping principle observed in semiconductors, when carbon is doped which has one less valence electron, one Li atom is removed to create a Li vacancy (VLi). This is analogous to p-type doping in semiconductors, whereby conditions conducive to the mobility of Li ions are created.32) Furthermore, when doping with oxygen, which has an additional valence electron, one Li atom was added to create interstitial Li (Lii). This is analogous to n-type doping in semiconductors, which had the effect of increasing the ion concentration along the Li ion diffusion pathways, thereby potentially enhancing ion mobility.33) Upon plotting the BVS for the materials where nitrogen atoms were doped with carbon and oxygen, as shown in Fig. 5, it was confirmed that all displayed a well-defined linear structure. To provide a more detailed comparison, the MSD was calculated, which demonstrated superior Li diffusion behavior in comparison to the undoped materials [Fig. 4(c-f)]. In particular, the O-doped LiPN2 structure exhibited good Li mobility, whereas it was relatively weak in the C-doped structure. This difference can be attributed to the electronic structure and bonding properties of O and C, which can act as a determining factor for ion mobility in the diffusion pathway of Li ions. Similarly, in the LiSi2N3 structure, the O-doped material exhibited better Li diffusion behavior than the C-doped material, although with considerably weaker movement overall. The additional Li contributes to a concerted ionic diffusion, resulting in improved kinetic performance compared to before.7,31) This suggests that oxygen doping appears to be more effective than carbon doping, supporting the idea that doping has a beneficial effect on Li diffusion. In our study, the O-doped LiPN2 material displayed the most active Li ion movement.
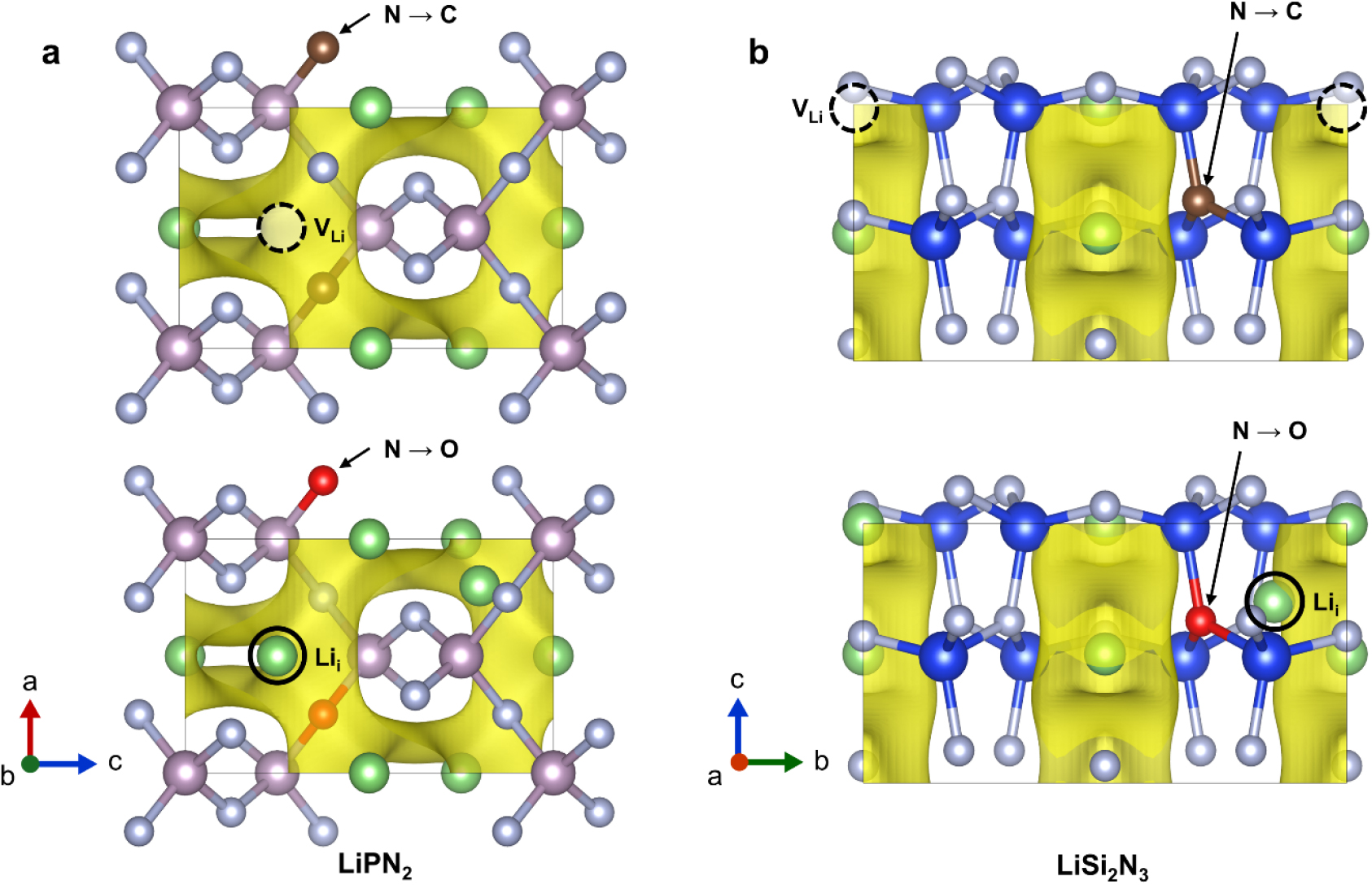
3.4. Li+ conductivity of O-doped materials
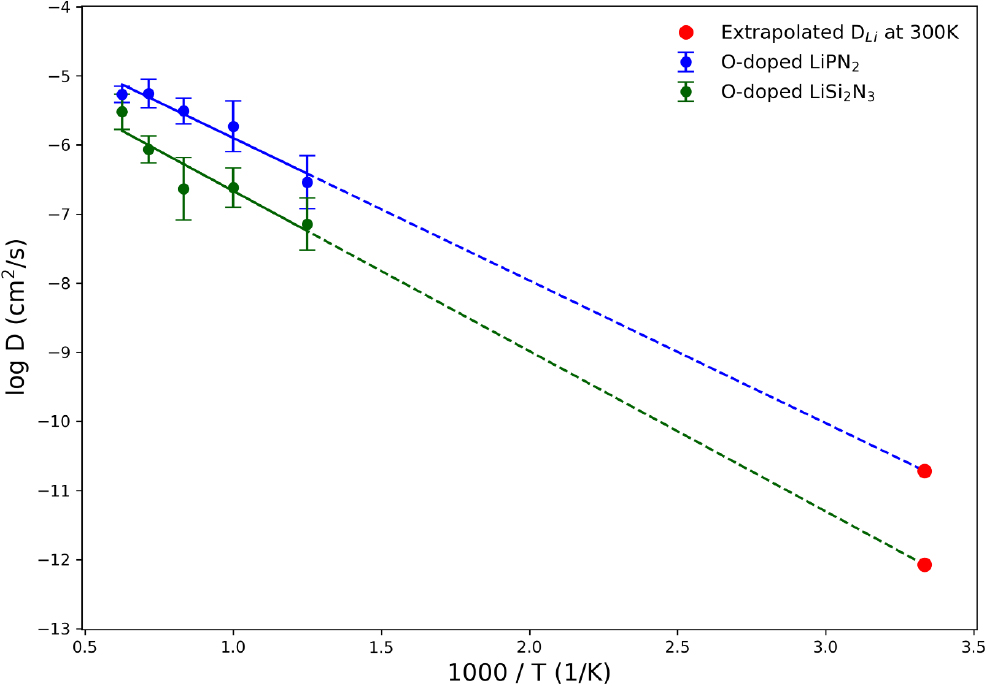
Fig. 6 shows the total average Arrhenius plot and the standard deviation of DLi for each temperature based on the AIMD results. The value of DLi at 300 K was extrapolated using the Arrhenius equation,
where, D0 and Ea are the pre-exponential factor and activation barrier, respectively. T and kB are temperature and Boltzmann constant, respectively. The extrapolated DLi at 300 K, obtained using the calculated D0 and Ea, was then employed to ascertain the ionic conductivity of the materials. Nernst-Einstein equation,
where, σ, n and z are the ionic conductivity, concentration of Li ions and the ionic charge number (for Li+, z = 1), respectively. The Faraday constant, diffusion coefficient, ideal gas constant and temperature are denoted by F, D, R, and T. Three additional independent ensemble AIMD simulations were performed (Fig. 7) for each material to establish the results in the preceding Fig. 4. The data obtained can contribute to optimizing the ionic conductivity properties of O-doped LiPN2 and O-doped LiSi2N3, and the consistency of the simulation results further improves the reliability of the theoretical predictions. In Fig. 6, the Arrhenius plot shows the comparison of O-doped LiPN2 and O-doped LiSi2N3 along with the extrapolated diffusion coefficient at 300 K. The activation energy (Ea) are 0.4096 and 0.4601 eV, with the diffusion coefficients at 300 K being 1.93 × 10-11 and 8.40 × 10-13 cm2/s for each respective material. For each temperature, the standard deviation was up to 2.6 × 106, with all values falling within this range. The ionic conductivities for O-doped LiPN2 and O-doped LiSi2N3, calculated using extrapolated values at 300 K, were σ = 4 × 10-3 and 11.2 × 10-5 mS/cm, respectively. This indicates that O-doped LiPN2 exhibits ionic conductivity properties about 100 times greater than O-doped LiSi2N3. Among the materials screened from the large-scale data, O-doped LiPN2 has been identified as the most suitable for a next-generation nitrogen-based SSE. Although this value still represents a conductivity that is too low for commercialization, it is expected that new materials, previously unknown, can be designed sufficiently by adjusting the aliovalent doping level. Solid electrolyte materials containing nitrogen have recently been reported experimentally.34,35) This suggests the possibility of proposing a different class of materials compared to oxides, sulfides, and halides, and further, it may allow for the suggestion of combined structures with other material classes.
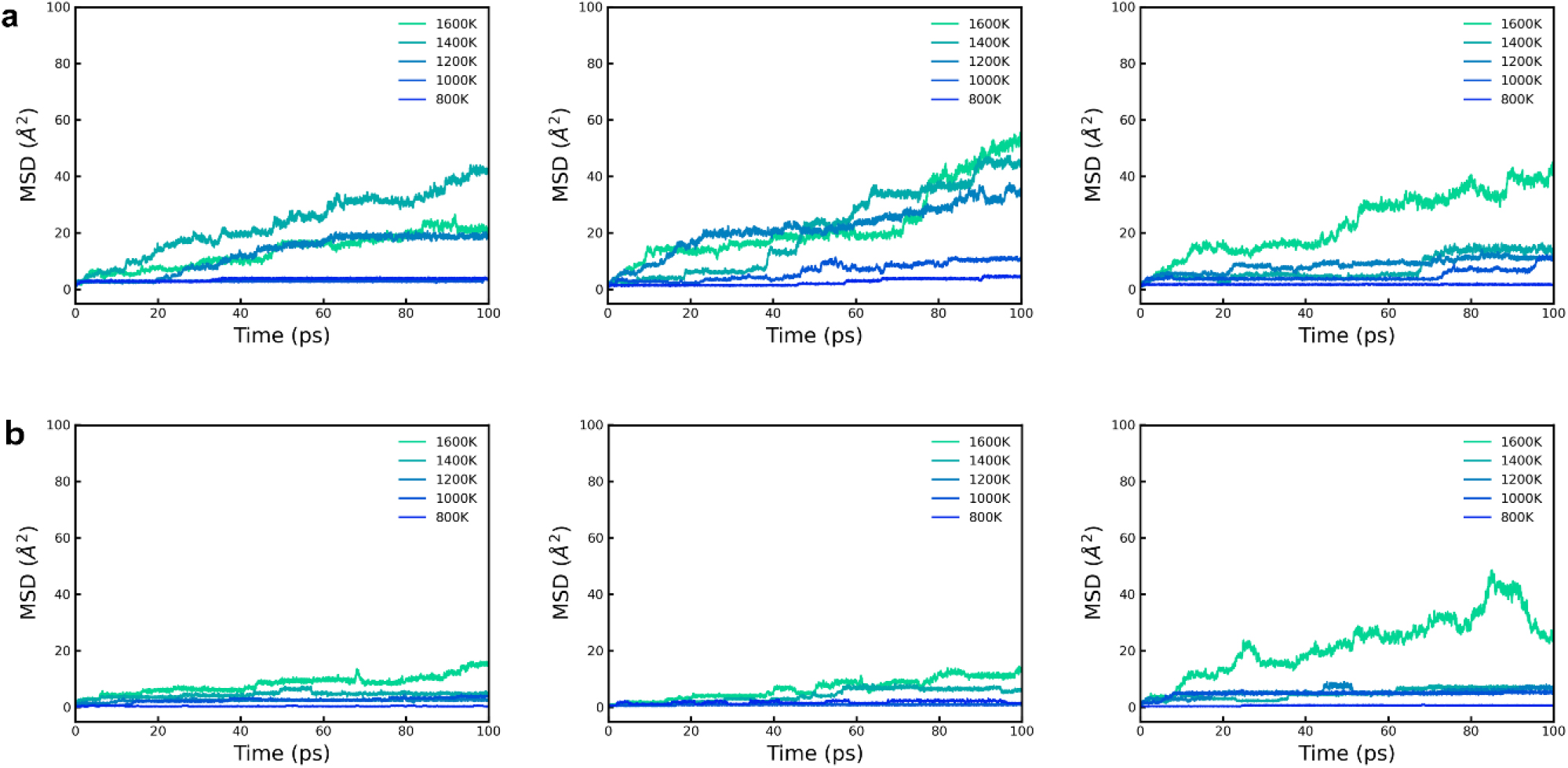
Fig. 7.
MSD plot of three ensemble structures. (a) O-doped LiPN2. (b) O-doped LiSi2N3. Due to the limited size of the original 1 × 1 × 1 unit cell, a larger 2 × 2 × 1 supercell was employed to ensure accurate representation of the structural and electronic properties during the AIMD calculations. This expansion allows for better accommodation of potential interactions and enhances the reliability of the simulation results.
4. Conclusion
We used the HTS technique to search for new nitrogen-based SSE materials. We found that LiPN2 and LiSi2N3 showed excellent Li diffusion pathways and were doped with oxygen and carbon for higher ionic conductivity. AIMD based on the ensemble structure resulted in ionic conductivities of 4 × 10-3 and 11.2 × 10-5 mS /cm for O-doped LiPN2 and O-doped LiSi2N3, respectively, and confirmed that O-doped LiPN2 outperformed O-doped LiSi2N3 by more than 100 times. Consequently, the O-doped LiPN2 material was found to be the most suitable SSE material. Through extensive analysis, we screened SSE materials based on several criteria and properties, and finally proposed one promising SSE material. Although our proposed material is not superior in terms of Li ion conductivity compared to currently researched and developed SSEs, this study is significant in that it provides an important methodology for exploring and screening new materials. In addition, the methodology proposed in this study is applicable not only to SSEs, but also to other battery materials and energy storage technologies and is expected to contribute to the development of more advanced materials. Our approach is expected to contribute significantly to the development of the next generation of energy storage technologies.
