

1. Introduction
Fluorine substituted conjugated organic molecules with donor-acceptor (D-A) architecture have garnered significant attention for development of efficient molecular materials for optoelectronic applications.1,2) The tuning of optoelectronic properties of the D-A systems has been achieved by incorporation of the F atom in place of H atom.3,4,5,6) Fluorine is the most electronegative element and acts as an inductive (σ) “electron-withdrawing group” (EWG).1,6) Upon incorporation in the D-A systems it perturbs the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) levels and improves the open-circuit voltage (Voc) in solar cells due to lowering of the HOMO level.6,7) Despite the considerable progress in organic solar cells (OSCs) technology, the quest for efficient donor materials still continues. With the advancement of computational chemistry it is possible to design and compare the theoretical results for the development of efficient D-A systems for solar cells.8,9,10) We were interested to explore the properties of fluorine-substituted bis-benzothiadiazole (FBBT) based systems for solar cell application. Our chosen molecular systems was based on one of the previous works reported on bis-benzothiadiazole (BBT) in which the H-atom at the 5 and 6 positions in the benzothiadiazole (BT) group were substituted by the F-atom.11)
BT group acts as a strong acceptor and has been widely functionalized and utilized to develop optoelectronic devices.12,13) The BT based molecular materials exhibit strong intermolecular interactions via heteroatom contacts and π-π interactions with well-ordered structures.12,13) BBT with two BT units can result in strong D-A interaction and lowering of HOMO-LUMO gap. The common strategies adopted to tune the properties of the conjugated organic systems involves: (a) substituting the donor/acceptor building blocks in the D-A systems, (b) grafting different π-linkers to extend the conjugation; and (c) incorporation of different functional groups in the D-A systems.5,14) We adopted the above listed strategies to tune the properties of FBBTs.
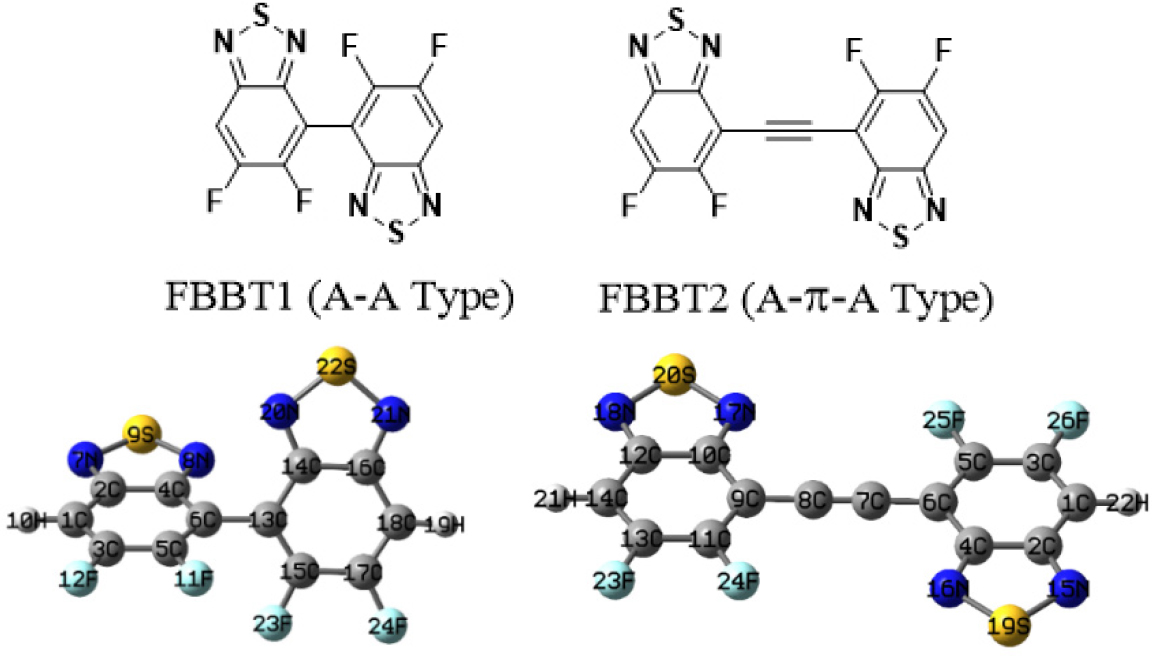
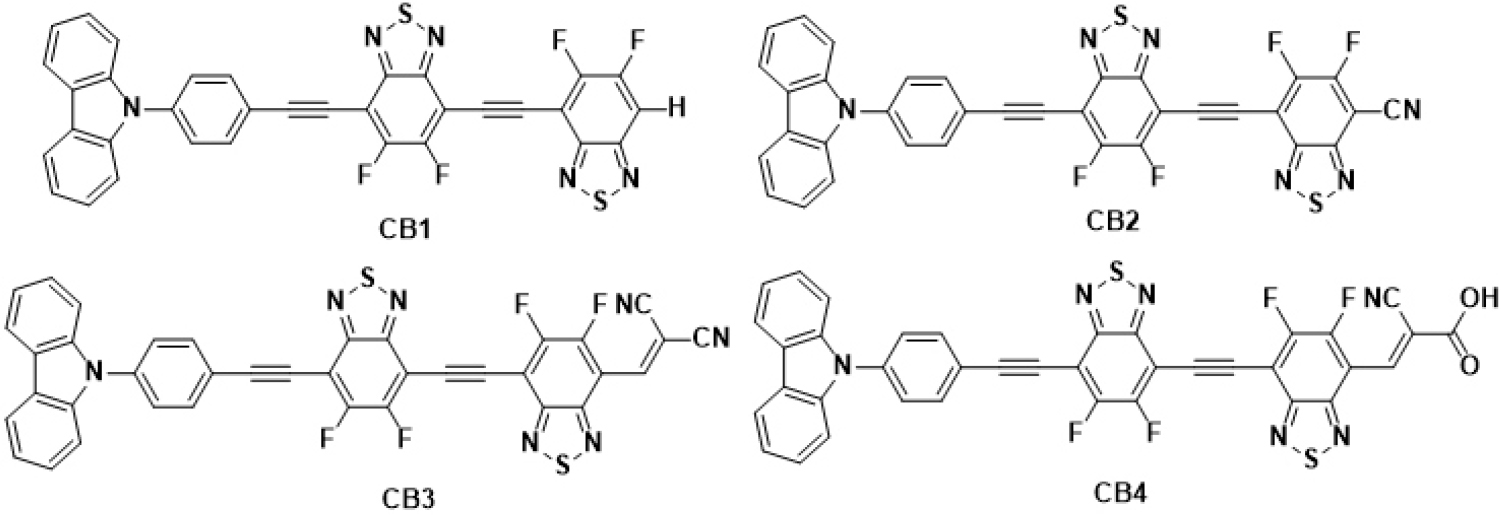
In the present study, we designed and theoretically studied D-A molecular systems of the type D-π1-A1-π2-A1-A2 based on FBBT. The design strategy involved replacing the H atom in BBT acceptor unit with F atom to obtain FBBT1 and FBBT2 (Fig. 1). The systematic tuning of optoelectronic properties of FBBT2 was achieved via π-extension and end-cap modeling. The computational studies show better planarity in FBBT2 compared to FBBT1 due to incorporation of π-extension between the fluorine-substituted benzothiadoazole acceptor units which is need for improved D-A interaction. The end-cap modeling was achieved by substitution of one of the H atom of FBBT2 with carbazole group to obtain CB1 as shown in Fig. 2. The heteroarene carbazole moiety was used as donor group due to its good hole transporting properties.15,16) The H atom on the acceptor FBBT2 in CB1 was further substituted by the acceptor cyano, dicyanovinyl and cyanoacrylic acid groups, to tune the optoelectronic properties of π-extended FBBTs17,18) The computational studies were performed using DFT and TDDFT methods to explore the optoelectronic properties of CB1-4. The results obtained exhibit systematic tuning of the HOMO and LUMO energy levels, HOMO-LUMO gap, and the photovoltaic properties. The fluorine substituted benzothiadiazole and cyano-acceptor end capped molecular systems have shown good film forming properties which indicates that the designed molecular systems with π-extended BBT with planar orientation, fluorine substitution and end-cap modeling with additional cyano acceptors can also exhibit good film forming properties.19,20,21) We feel the present study could assist to develop efficient use FBBT based small molecular materials for bulk heterojunction (BHJ) solar cells.
2. Experimental Procedure
The FBBT1-2 and end-capped FBBTs CB1-4 were optimized utilizing the density functional theory (DFT) with Becke’s three-parameter functional and Lee-Yang-Parr functional (B3LYP) combined with 6-31G(d,p) basis set.22,23,24,25) The TDDFT calculations using functional MPW1PW91 (spin restricted parameters) and 6-31G(d,p) basis set were performed with the IEFPCM solvation method employing chloroform solvent medium.10,22,25) All the calculation were performed using Gaussian 09 package and GaussView 5.0 is utilized for visualization of the results and obtaining data from the output.22,23,24,25)
3. Results and Discussion
3.1. Optimization of the geometry
Optoelectronic properties of D-A molecular scaffolds are closely interrelated to their ground state geometry.8,10) Therefore to study the features of these D-A molecular scaffolds we investigated their optimized ground state geometry to explore the bond parameters and electronic structures which were computed through DFT method at B3LYP level.8,9,22)
The optimized structure of FBBT1 and FBBT2 is shown in Fig. 1. The FBBT1 has a non-planar arrangement with dihedral angles of 57.7° between the fluorine-substituted benzothiadiazole units which is similar to that observed in single crystal structure of BBTs.11) The planar orientation of the acceptor benzothiadiazoles is desirable for strong interaction in D-A systems. Hence, we designed FBBT2 with extended π-conjugation by incorporation of ethyne linker between the fluorine-substituted benzothiadiazole units.26) The optimized structures of FBBT2 shows planar arrangement which can result in strong intramolecular charge transfer (ICT) upon end-cap modeling.
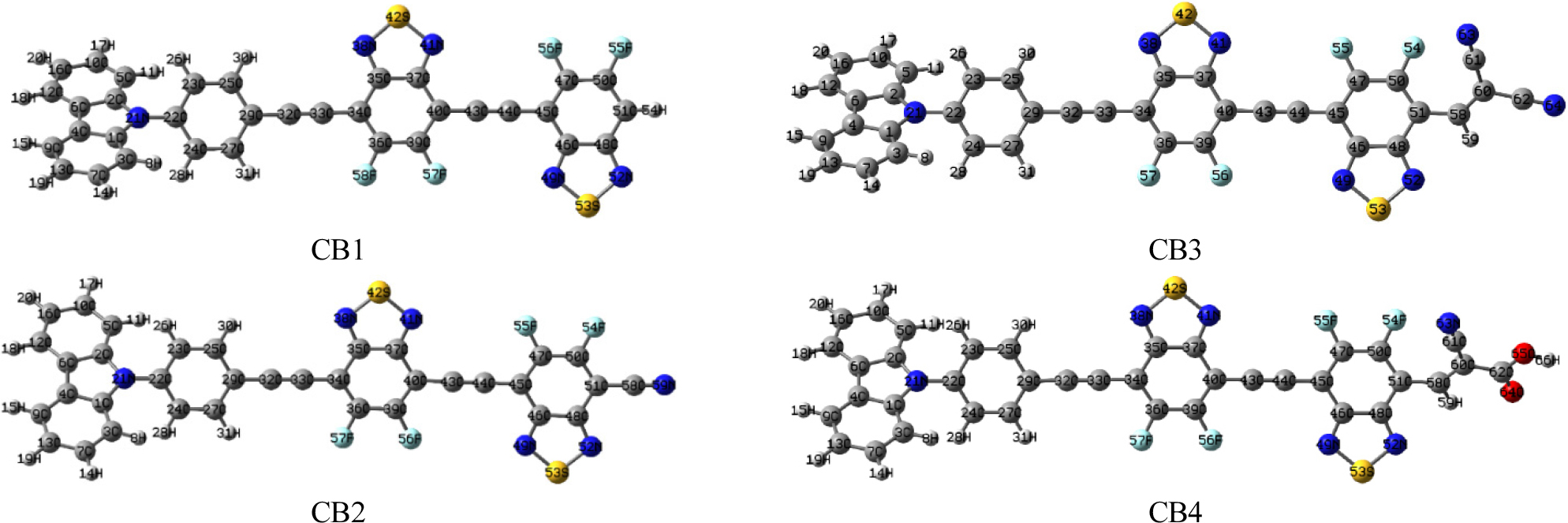
Based on the above results FBBT2 was considered for end-cap modeling via incorporation of carbazole and cyano acceptor groups (cyano, dicyanovinyl and cyanoacrylic acid groups) to tune the optoelectronic properties (Fig. 2).18,19) The optimized structures of CB1-4 are presented in Fig. 3. The carbon-carbon (C-C) bond length in these D-A scaffolds (1.41 Å to 1.42 Å) ranges between that of C-C single bond (1.54 Å) and C-C double bond (1.34 Å) which indicates the presence of π-conjugation via π-electron delocalization, as required for improved charge transport characteristics. The lower bond length of the D-A molecular scaffolds shows better conjugation.27,28) The dihedral angle between the donor carbazole moiety and the adjacent phenyl ring with the FBBT was examined. The carbazole donor in CB1-2 shows non-planar arrangement, whereas the phenyl ring and the FBBT acceptor shows planar orientation which supports conjugation and better charge transfer (CT).22,28)
3.2. Frontier molecular orbitals and electronic properties
The electronic properties of molecular materials rely on its frontier molecular orbitals (FMOs).8,10,22,29) In this regard the donor-acceptor strategy has received significant attention as they offer tuning of FMOs for development of efficient molecular materials.13) An initial insight to establish the candidature of D-A molecular scaffolds for OSCs is provided by computational studies to probe the HOMO and LUMO energies.22,29,30) To study the effect of systematic extension of π-conjugation and sequential functionalization with carbazole and cyano-based acceptor groups on the properties of FBBTs, we calculated the HOMO and LUMO orbital energy values and the HOMO-LUMO Gap, as shown in Fig. 4 and Fig. 5.
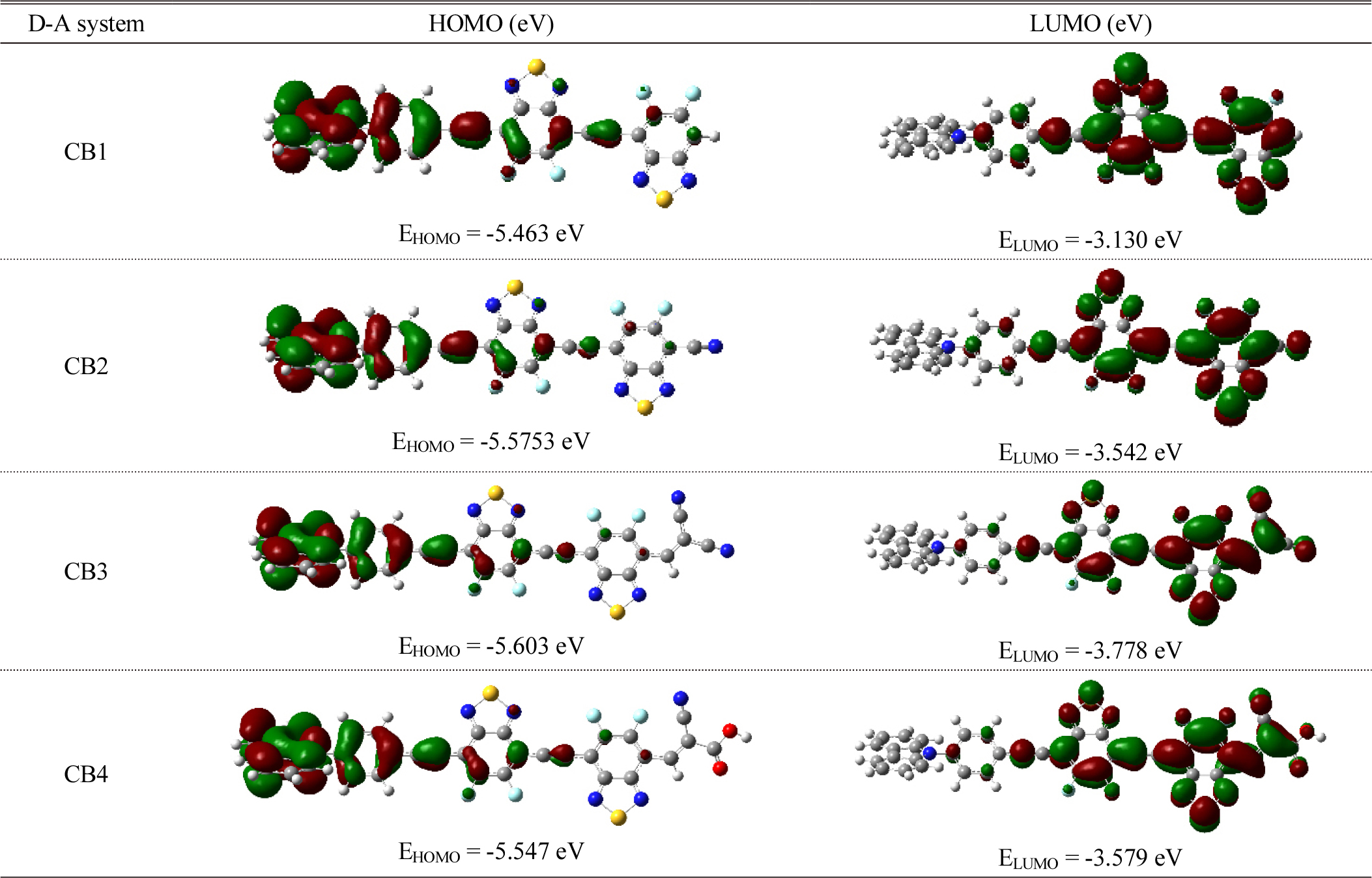
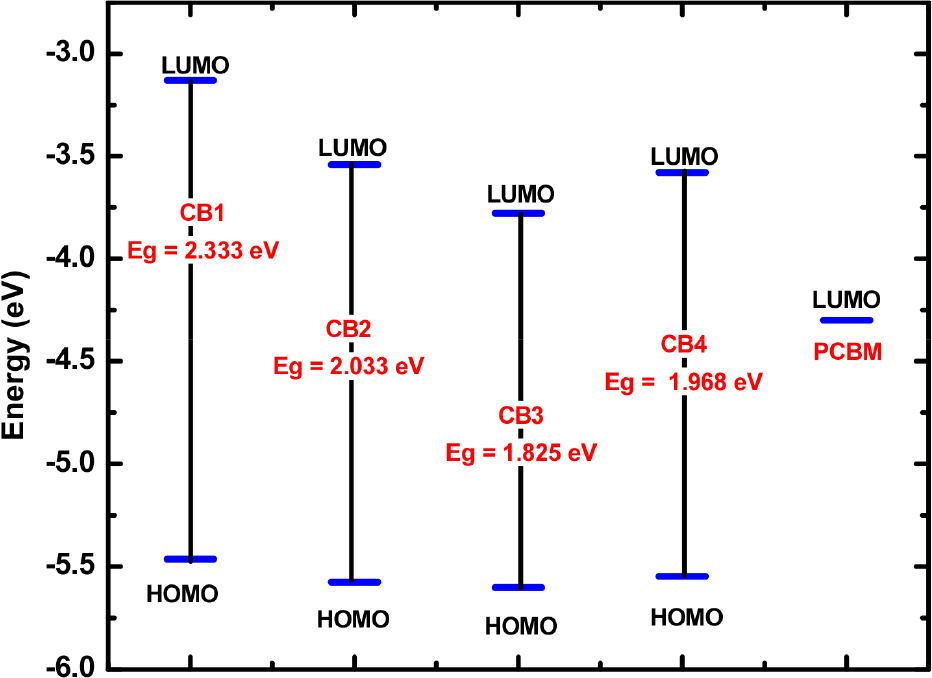
Based on the results obtained, we found that: (i) The LUMOs in these D-A scaffolds CB1-4 are delocalized on FBBT and extending over the cyano-based acceptor groups in CB2-4. The extension of delocalization of LUMO over CN acceptor groups in CB2-4 results in greater stabilization of the LUMO levels. (ii) The HOMOs in these D-A scaffolds CB1-4 are delocalized on the carbazole donor moiety and adjacent phenyl ring. (iii) The designed D-A scaffolds exhibit the HOMO-LUMO gap in the range from 2.333 eV to 1.825 eV. The computed HOMO-LUMO gap follows the trend CB1 > CB2 > CB4 > CB3. The trend shows that the end-cap modeling FBBT results in lower HOMO-LUMO gap owing to stronger electron-withdrawing nature of cyano-based acceptor groups.18,19)
3.3. Photovoltaic properties
OSCs are composed of a mixture of p-type (donor material) and n-type semiconducting components (acceptor material).22,31) The power conversion efficiency (PCE) of these OSCs can be improved by tuning the common factors such as lowering of the HOMO-LUMO gap and increasing the open-circuit voltage (Voc), Jsc (short-circuit current density), and the fill factor (FF). The PCE can be calculated according to the Eq. (1) given below:22,31)
Where, Pinc (incident photon to current efficiency). The purpose of this study is to explore the effect of sequential substitution of carbazole and cyano-based acceptor groups to tune the photovoltaic properties of π-extended FBBT based D-A systems. To explore the candidature of these D-A systems we compared their Voc and PCE. The Voc of OSCs can be computed from the difference between the HOMO of donor (CB1-4) and LUMO of acceptor (PC71BM), taking into account the energy lost during the photo-charge generation.22,31) The calculated Voc values have been obtained using the following Eq. (2):22,24,31)
Here, (e) is the elementary charge, the LUMO energy level of PCBM is -4.3 eV, and a value of 0.3 V is used as the empirical factor.
Table 1.
Energy values of ELUMO, EHOMO and the open circuit voltage (Voc), LD-LA, PCE and fill factor (FF) of the studied molecules CB1-4 obtained by B3LYP/6-31G(d,p) level.
The computed values of Voc for CB1-4 with PC71BM acceptor are listed in Table 1. The Voc value exhibits the trend CB3 > CB2 > CB4 > CB1. Based on the results obtained, we found that sequential incorporation of cyano acceptor group leads to higher Voc in CB2-4 due to stabilization of HOMO orbital.18,22) The Voc parameter of FBBT D-A systems can be tuned efficiently by increasing the acceptor strength via end group modeling. Table 1 shows the LUMO energy level difference (LD-LA) between the FBBT based donor CB1-4 and the PC71BM acceptors. The calculated values of (LD-LA) are suitable for efficient electron injection from the donor to the LUMO of the PC71BM acceptor.8,22) The results show that the studied FBBT based donor CB1-4 can be utilized as efficient donors for BHJ solar cells.22,24,31) To achieve high PCE, the donor should exhibit large FF values. The FF values for FBBTs CB1-4 were calculated and compiled in Table 1. FF is using Eq. (3) as:27,28)
with, 𝜈oc denoting dimensionless voltage and calculated using Eq. (4)
The results show that the FF are influenced only slightly upon cyano group incorporation. The PCE values using the Scharber diagram for OSCs based on PC71BM acceptor is tabulated in Table 1.31) The designed molecules CB1-4 show the theoretical PCE between ~3.0% to ~8.0%. CB3 exhibits the highest PCE value which can be attributed to the strong electron-withdrawing nature of the dicyanovinyl group.18,19) The results illustrate that end-cap modeling of FBBTs with cyano-based acceptor groups can significantly enhance the solar cell efficiency of π-extended FBBTs and the designed molecules with sufficiently high Voc and FF values can serve as good candidates for organic BHJ solar cells.
3.4. Optical Properties
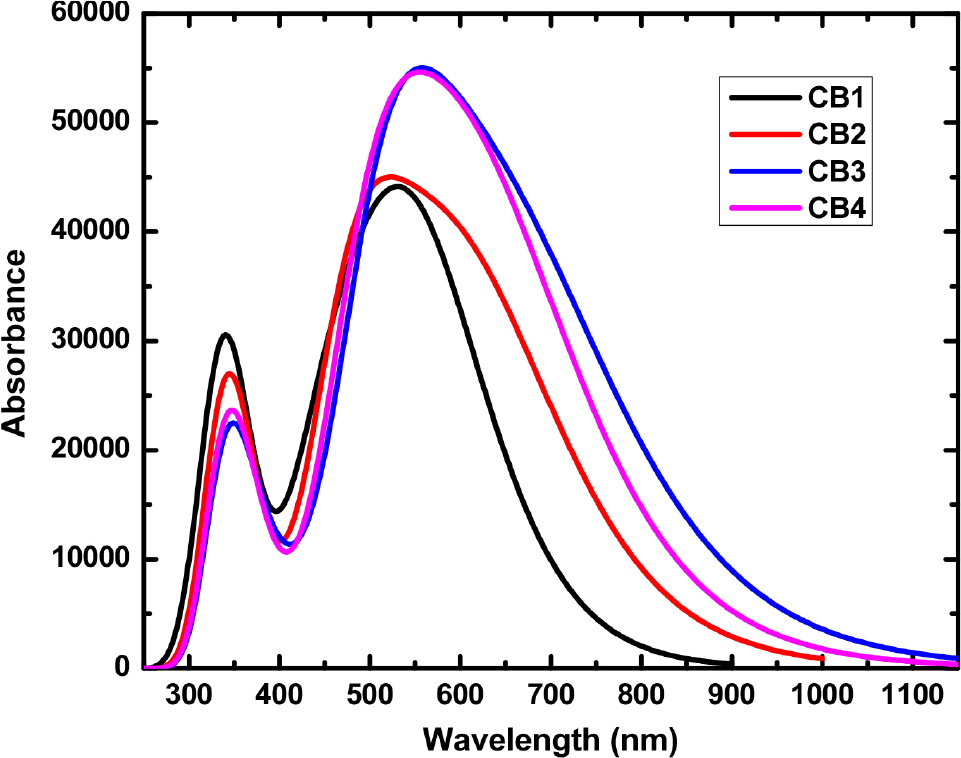
The optical properties of D-A scaffolds CB1-4 were studied using, time-dependent density functional theory (TDDFT) method. The TDDFT calculations were performed using the MPW1PW91 functional at the 6-31G (d,p) level with the IEFPCM solvation method employing chloroform.10) The calculated maximum absorption wavelength (lmax), oscillator strength (ƒ) and vertical excitation energies (Eex) for chloroform solvent is included in Table 2.22) The molecules under investigation exhibit strong absorption in the visible region as shown in Fig. 6. The incorporation of cyano-based acceptor group in CB2-4 results in significant red-shift in the absorption maxima compared to CB1. This could be attributed to the extended conjugation and stronger electron-withdrawing character of FBBT with terminal cyano-based acceptor groups.18,19) The λmax values refer to electronic transitions involving the electron transfer from HOMOs located over the donor carbazole unit to LUMOs delocalized over the fluorine-based bis-benzothiadizole acceptor and extending over the cyano-based acceptor groups in CB2-4. The TDDFT studies indicate significant tuning of optical properties by end-cap modeling of FBBTs.
Table 2.
Calculated maximum absorption wavelengths λmax (nm) in chloroform, electronic transition energy Eex (eV), oscillator strength f (au), and main electronic transitions, calculated electronic absorption spectra for CB1-4.
4. Conclusion
In summary, FBBTs were designed and investigated using DFT and TDDFT methods. The incorporation of π-extension induced better planarity in between the neighboring FBBT acceptor units which is required for strongly interacting donor-acceptor (D-A) systems and efficient ITC. The optoelectronic properties of FBBTs were further tuned by end-cap modeling using donor and acceptor groups. The computational results reveal that the donor carbazole fragment adopts a non-planar configuration, while the remaining linkers and acceptor fragments show a good planarity favorable for limiting π-π aggregation. These D-A scaffolds show HOMOs delocalized on donor carbazole, and LUMOs delocalized at the acceptor FBBT and the cyano-based acceptor groups. The Voc trend CB3 > CB2 > CB4 > CB1 shows that incorporation of cyano based acceptor groups can lead to better Voc in these FBBT. TDDFT calculation results indicate that incorporation of cyano-groups enhances the acceptor strength and can result in stronger absorption and red shift of absorption spectra. Overall, this study shows that π-extension between the acceptor FBBT units together with end-cap modeling with a strong donor/acceptor groups can be a promising approach to systematically tune the optoelectronic and photovoltaic properties.
