

1. Introduction
Miniaturization of high capacitance multilayer ceramic capacitors (MLCCs) is one of the major concerns in development of microelectronics for IoT and smart mobility applications. For that, thin dielectric layers with large capacitance are desired. These extremely high-level compactness of the dielectric layers in MLCC can only be achieved through high crystallinity of nano-scale ferroelectric ceramic powders and their microstructure control upon sintering procedure. Anticipated cutting-edge premium MLCC incorporates ultra-thin barium titanate (BT) ferroelectric layers with a thickness of approximately 1~2 µm, exhibiting an exceptionally refined nano-scale microstructure that enhances the performance and reliability of electronic devices. Thus, synthesis of ultrafine BT powders with particle size smaller than 100 nm and with high crystallinity and tetragonality (defined as c/a, the relative ratio of the BaTiO3 unit cell lattice parameters of c- to a-axis) is a critical must-have technology to achieve further compactness of the high-end MLCCs.1,2,3)
The conventional synthesis method for the BT powder is represented by mixed oxide route, which is based on the formation of the perovskite phase by a diffusion controlled the solid-state reaction of barium carbonate and titanium oxide mixture at high temperatures over 1,100 °C under specific pH and atmospheric conditions.4) Such high calcination temperatures lead to aggregated coarse powders that need to be separated by post milling process. To synthesize high purity nano-sized fine BaTiO3 particles, solution-based processes such as the oxalate route, co-precipitation, sol-gel method, hydrothermal synthesis, etc. were widely used.5,6,7,8,9) Especially, the hydrothermal synthesis is one of the most suitable processes to obtain highly crystalline BaTiO3 nanoparticles at low temperatures by an environmentally friendly way.
For the hydrothermal synthesis of BaTiO3, various starting materials as titanium sources are available. In the early days, hydrolysis of titanium dioxide (TiO2) particles followed by the dissolution-reprecipitation reaction was mainly applied. Anatase phase TiO2 was mainly used for synthesizing high crystallinity nano-sized BaTiO3 by controlling variables such as Ba/Ti ratio, pH, time, temperature, etc.10,11,12) However, the uncontrolled particle size of the precursor material limited the production of homogeneous and sufficiently fine BT powder. As ultrafine TiO2 powder became available in the market in the early 2000s, some researchers started to utilize the commercial TiO2 nanopowder (70 % anatase and 30 % rutile, Degussa P25) with a size of ~30 nm instead of the conventional anatase TiO2. The ultrafine TiO2 with controlled particle size and morphology attributed to the production of BaTiO3 with a uniform size of less than 100 nm at low temperatures in a short reaction time. The large surface area of the nanoparticle precursors accelerated the dissolution rate and played a critical role in determining the crystal size of the BT product. However, the resulting BT product were in cubic perovskite phase which had to be annealed to transform into ferroelectric tetragonal phase.13,14,15) TiO2 nano-sols are also widely used Ti source materials for ultrafine BT nanoparticle synthesis. The hydrothermal synthesis utilizing TiO2 nano-sols could produce BT nanoparticles (d ≈ 100 nm) with high tetragonality after subsequent annealing procedures at elevated temperatures.16,17) Various titanium-alkoxide precursors have also been used as a titanium source in hydrothermal synthesis. Titanium (IV) isopropoxide (Ti{OCH(CH3)2}4) and tetrabutyl titanate (Ti(C4H9O)4) precursors were very effective in producing uniform BT nanoparticles smaller than 100 nm. However, the as-synthesized BT particles were mostly in lack of tetragonal symmetry, thus subsequent annealing is necessary to achieve the high tetragonality.18,19,20)
As a barium source material, although barium hydroxide (Ba(OH)2) has been mostly used for the hydrothermal synthesis in strong basic condition (pH > 13), various types of starting Ba-salts including halides, nitrates and acetates are still available, and the barium source materials for the BT hydrothermal synthesis are also known to influence the particle size and tetragonality of the product particles due to the solubility changes in a specific condition.21)
In this study, we revisit the TiO2-to-BT hydrothermal conversion using economical, widely available precursors to identify a practical route to highly tetragonal BT nanoparticles without post-annealing. Commercial TiO2 (P25) was adopted as the Ti source, while barium acetate—owing to its high aqueous solubility—was compared directly with Ba(OH)2 to assess impacts on phase purity (notably BaCO3 formation), particle-size distribution, and tetragonality. We also scrutinize the role of Ba/Ti ratio on nucleation and growth, demonstrating that controlled Ba/Ti chemistry can simultaneously narrow particle size and enhance tetragonality; importantly, the results confirm that TiO2 remains a competitive and scalable precursor for BT nanoparticle synthesis under optimized hydrothermal conditions.
2. Experimental Procedure
BaTiO3 nanoparticles were prepared hydrothermally using barium acetate (Ba(CH3COO)2, ≥ 99 %, Sigma-Aldrich/Merck; CAS 543-80-8) or barium hydroxide octahydrate (Ba(OH)2・8H2O, ≥ 98 %, Sigma-Aldrich/Merck; CAS 12230-71-6) as Ba sources, TiO2 (P25, Evonik-Degussa; CAS 13463-67-7) as the Ti source, and potassium hydroxide (KOH pellets, Duksan; CAS 1310-58-3) as the mineralizer. The required amounts of raw materials and 25 mL of distilled water were charged into a Teflon vessel and stirred for 30 min to disperse TiO2. The TiO2 concentration was fixed at 0.02 M, and the Ba/Ti molar ratio was varied from 1.0 to 2.0. After stirring, 25 mL of 6 M KOH was added. The suspension was transferred to a stainless-steel autoclave and reacted at 240 °C for 6 h. The product was collected and washed five times with distilled water by centrifugation (8,000 rpm, 15 min).
For residual carbonate removal, selected batches were additionally washed three times with the dilute solutions (0.5~1.0 M) of each acids: nitric acid (HNO3, 70 %, Daejung; CAS 7697-37-2), citric acid (C6H8O7, anhydrous, Sigma-Aldrich; CAS 77-92-9), or acetic acid (CH3COOH, glacial, Daejung; CAS 64-19-7). The washed powders were dried in an oven at 120 °C for 10 h and gently ground in an agate mortar.
Phase identification was performed by X-ray diffraction (XRD, Ultima IV, Rigaku Co., Japan) using Cu Kα radiation (λ = 1.5418 Å). Particle morphology and size were examined by field-emission scanning electron microscopy (FE-SEM, JEOL-7800F, JEOL Ltd., Japan). Raman spectra were collected on a LabRam Aramis spectrometer (Horiba Jobin Yvon, France) over 100-1,000 cm-1. FT-IR spectra were obtained on a Vertex 70 (Bruker Co., Germany) over 400-4,000 cm-1.
3. Results and Discussion
Fig. 1 shows how the phase assemblages change when the Ba source is changed but the hydrothermal conditions stay the same. The reflections indexed to BaTiO3 match the tetragonal perovskite in both cases. The (002)/(200) peak splitting near 2θ ≈ 45° shows that the symmetry is tetragonal, not cubic.
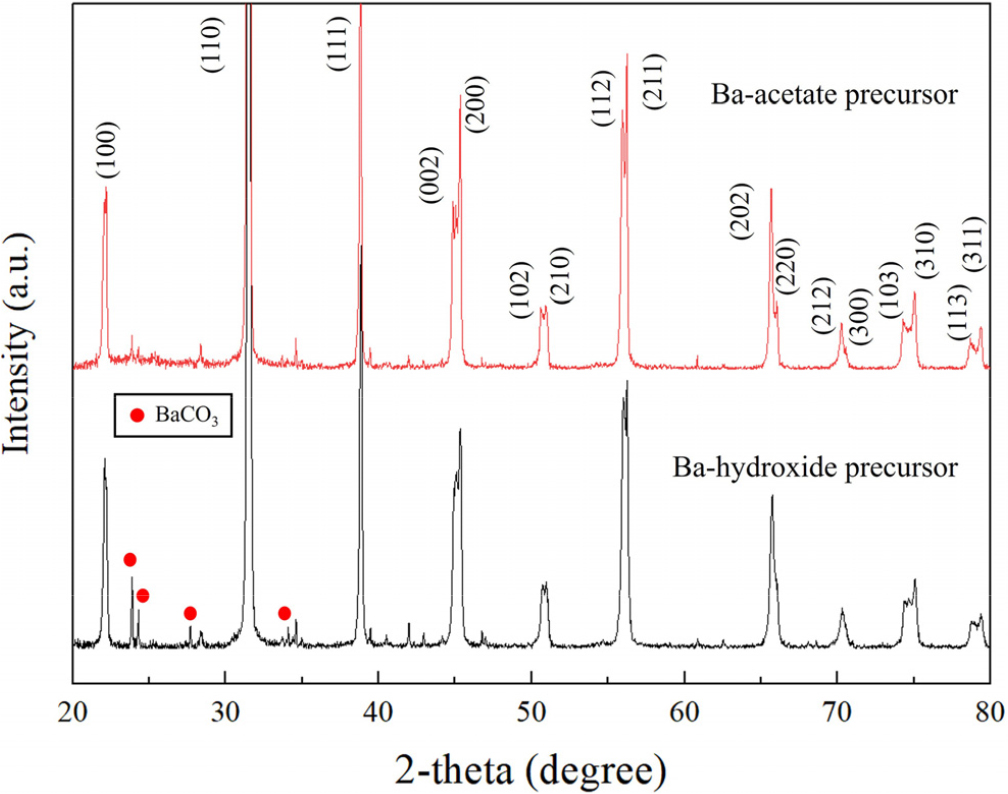
The Ba(OH)2 route shows extra reflections (red dots in Fig. 1) which comes from the BaCO3 secondary phase, while the Ba(CH3COO)2 route lowers these carbonate peaks to near the detection limit. This difference is in line with how precursor chemistry affects dissolution and the formation of secondary phases. It is well established that strongly alkaline solutions (NaOH, KOH, metal hydroxides) exhibit a greatly enhanced effective solubility for CO2 due to rapid chemisorption and conversion into carbonate and bicarbonate species; at pH ≳ 10, this reactive absorption allows substantial CO2 uptake and, in the presence of divalent cations such as Ba2+, leads directly to precipitation of carbonates (e.g., BaCO3, CaCO3).22) When Ba(OH)2 is employed as the Ba source, the hydrothermal reaction proceeds in a strongly alkaline (pH ≳ 13) environment. Under these conditions, dissolved atmospheric CO2 is quantitatively converted to CO32-, and the high activity of free Ba2+ favors precipitation of BaCO3 as an impurity phase. In addition, unreacted Ba(OH)2 or BaO remaining on the surface of BaTiO3 particles readily carbonizes during washing and drying, further increasing the BaCO3 content detected ex-situ. In contrast, Ba(CH3COO)2 produces only relatively mild basic solutions, in which the carbonate species are dominated by HCO3- and the concentration of CO32- is significantly lower; partial complexation of Ba2+ by acetate additionally decreases the free Ba2+ activity. As a result, the Ba-acetate system exhibits a much weaker driving force for BaCO3 precipitation, and stoichiometric Ba-Ti acetate gels can be hydrothermally converted to BaTiO3 with nearly quantitative yield and without using Ba excess, typically yielding powders in which BaCO3 is not detected by XRD.23,24) Consequently, BaTiO3 powders synthesized from Ba(OH)2 generally contain more BaCO3 than those obtained from Ba(CH3COO)2 under comparable conditions, particularly when a Ba excess and very high pH are used to promote tetragonal phase formation. We also emphasize that the tetragonal signatures are already present without post-annealing, and that carbonate formation is the principal differentiator between the two Ba sources under these conditions. The acid-washing study in the next sections shows that mild acids can effectively remove any trace carbonate that is left over after the acetate route, which makes the phase even cleaner.
Fig. 2 shows how morphology changes with the Ba/Ti ratio for the barium acetate route (panels a-e). The red-boxed panel shows a sample made using the Ba(OH)2 route so that it can be directly compared to the same Ba/Ti = 2.0 condition. As the Ba/Ti ratio rises along the acetate route, the average particle size diminishes and the distribution narrows; for Ba/Ti ≥ 1.5, the powders consist mainly of ~80-120 nm particles exhibiting mixed spherical and faceted morphologies, signifying growth at the size where surface-energy minimization and crystallographic faceting equilibrate.

The Ba(OH)2 sample in the red box, on the other hand, has coarser, less uniform particles, with some grains that are too big and a wider range of sizes than the acetate sample at Ba/Ti = 2.0 (see red box vs. panel e). This difference fits with the way hydrothermal BT forms: Ti-O bonds break down into [Ti(OH)x]4-x species, and BT forms with Ba-bearing species (Ba2+, BaOH+). When Ba(OH)2 is used as the Ba source, it doesn’t dissolve completely, which lowers the free Ba concentration in the solution. This causes inhomogeneous nucleation and runaway grain growth in areas where Ba is plentiful, which is why there are a lot of different sizes and sometimes big particles.
The acetate route works better because it gives Ba a higher, more even activity in solution and stops the formation of BaCO3 compared to Ba(OH)2, as shown by XRD (Fig. 1). This combination increases the effective nucleation density and slows down secondary ripening. This results in nanoparticles that are smaller and more evenly distributed at a given temperature and dwell time. The trend of increasing Ba/Ti from 1.0 to ≥1.5 progressively tightens size distribution is due to (i) better Ba availability during the critical early nucleation window and (ii) less Ba scavenging into secondary carbonate phases. Both of these effects are stronger for Ba(CH3COO)2 than for Ba(OH)2 under the same conditions. It is confirmed that controlling Ba/Ti in the acetate route is an effective way to consistently get uniform BT nanoparticles that are about 100 nm in size. In contrast, the Ba(OH)2 route (red box) tends to give particles that are wider and less controlled in size when using the same hydrothermal schedule.
Fig. 3 summarizes the phase and symmetry evolution of hydrothermally synthesized BaTiO3 (240 °C, 6 h) as a function of Ba/Ti ratio. Across all compositions, the powders exhibit the tetragonal phase at room temperature, as confirmed by the clear (002)/(200) peak splitting near 2θ ≈ 45° and the calculated tetragonality [Fig. 3(a)]. As Ba/Ti increases from 1.5 to 1.7, the tetragonality increases and reaches a maximum at Ba/Ti = 1.7 (c/a = 1.007615). With further Ba/Ti increase (≥ 2.0), the tetragonality gradually decreases, consistent with the modest reduction in the (002)/(200) separation in Fig. 3(a). These results align with prior reports that hydrothermal BaTiO3 often appears pseudo-cubic due to OH- defects and oxygen vacancies, which are mitigated by using excess Ba to compensate Ba deficiency during nucleation and growth.25,26,27) In our case, moderate Ba excess improves tetragonality up to Ba/Ti = 1.7, but larger excess can be counterproductive. One possible reason is that secondary phases (like BaCO3) can form in Ba-rich, strongly alkaline environments. These phases can take Ba2+ out of solution and cause local Ba deficiency again during perovskite growth. This explains why tetragonality goes down after the best value.19,25,26,27)
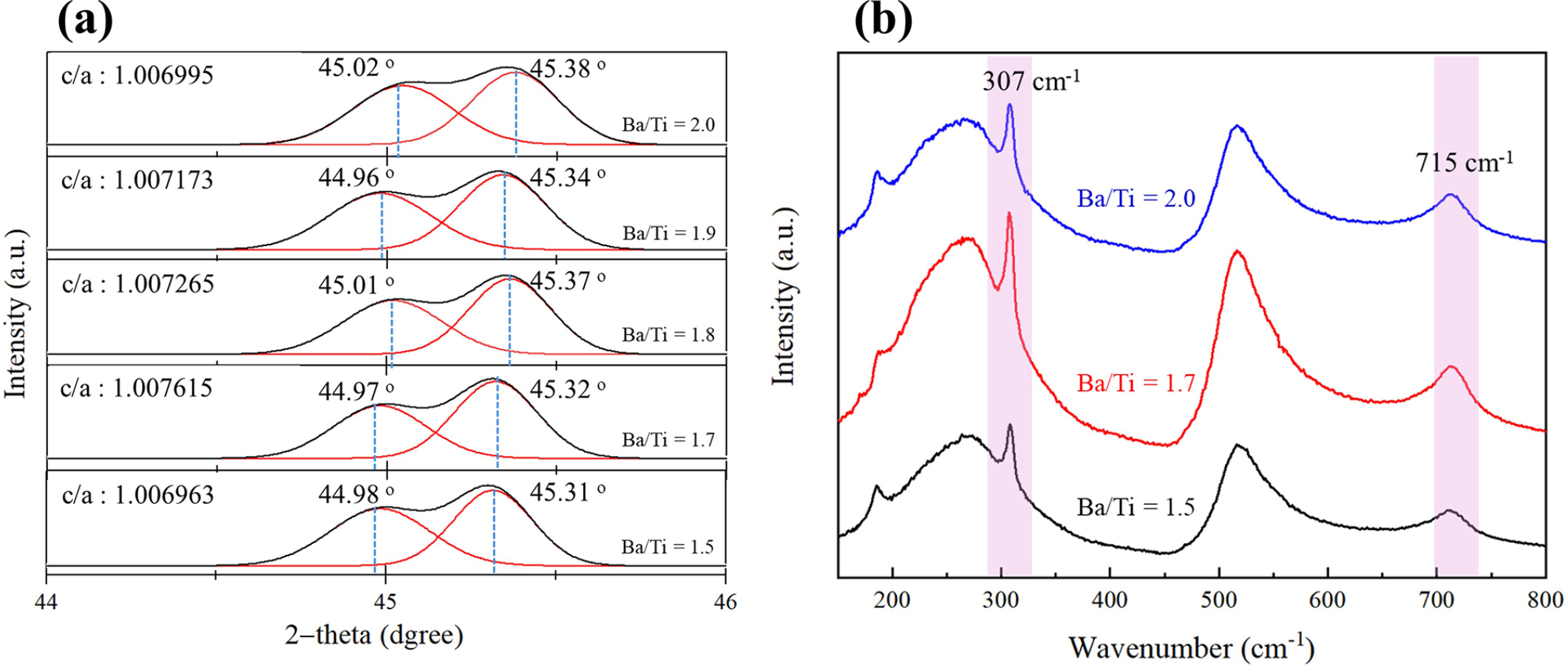
Fig. 3.
(a) X-ray diffraction peaks in the 44°~46° two-theta range (b) Raman spectra of the as received BaTiO3 nanoparticles prepared by the Ba(CH3COO)2 route hydrothermal synthesis with various Ba/Ti ratio; the red lines in (a) indicate Gausian fitting lines for each (002) and (200) diffraction peaks.
Because XRD-derived tetragonality can be difficult to quantify reliably for nanosized powders—peak broadening and microstrain can obscure (002)/(200) separation even after Rietveld refinement27)—we employed Raman spectroscopy as a complementary probe of symmetry [Fig. 3(b)].27,28,29,30) All samples show Raman bands near 185, 250, 307, 515, and 715 cm-1, consistent with BaTiO3. The assignments for the tetragonal phase are: A₁(TO) ≈ 250 cm-1, B₁/E(TO+LO) ≈ 307 cm-1, [E(TO), A₁(LO)] ≈ 515 cm-1, and [E(LO), A₁(LO)] ≈ 715 cm-1 [27,29]. Since both tetragonal and cubic BaTiO3 can exhibit bands near 185, 250, and 515 cm-1,27) the 307 and 715 cm-1 bands are most diagnostic for tetragonal symmetry; their sharpness/intensity can be used to compare relative tetragonality among samples.30) In our series, the 307 cm-1 band is sharpest and most intense at Ba/Ti = 1.7, whereas reduced tetragonality (Ba/Ti = 1.5 or ≥ 2.0) correlates with broader and slightly weaker features—fully consistent with the XRD trend.27,28,29,30) Taken together, XRD and Raman indicate that Ba/Ti ≈ 1.7 is an optimal window where Ba-deficiency-related defects are minimized, leading to the highest room-temperature tetragonality, while excessive Ba promotes secondary-phase formation (e.g., BaCO3) that can degrade crystallinity and effective stoichiometry.19,25,26,27,28,29,30)
Finally, we note that some studies report a ~3 nm cubic shell surrounding hydrothermal BaTiO3 nanoparticles, an effect that becomes more pronounced with decreasing size and could subtly bias XRD-based symmetry evaluation toward more cubic-like signatures even when local tetragonal order persists.26,27) This possibility further underscores the value of conjoint XRD-Raman analysis for reliable assessment of tetragonality in hydrothermal BaTiO3 nanopowders.26,27,28,29,30)
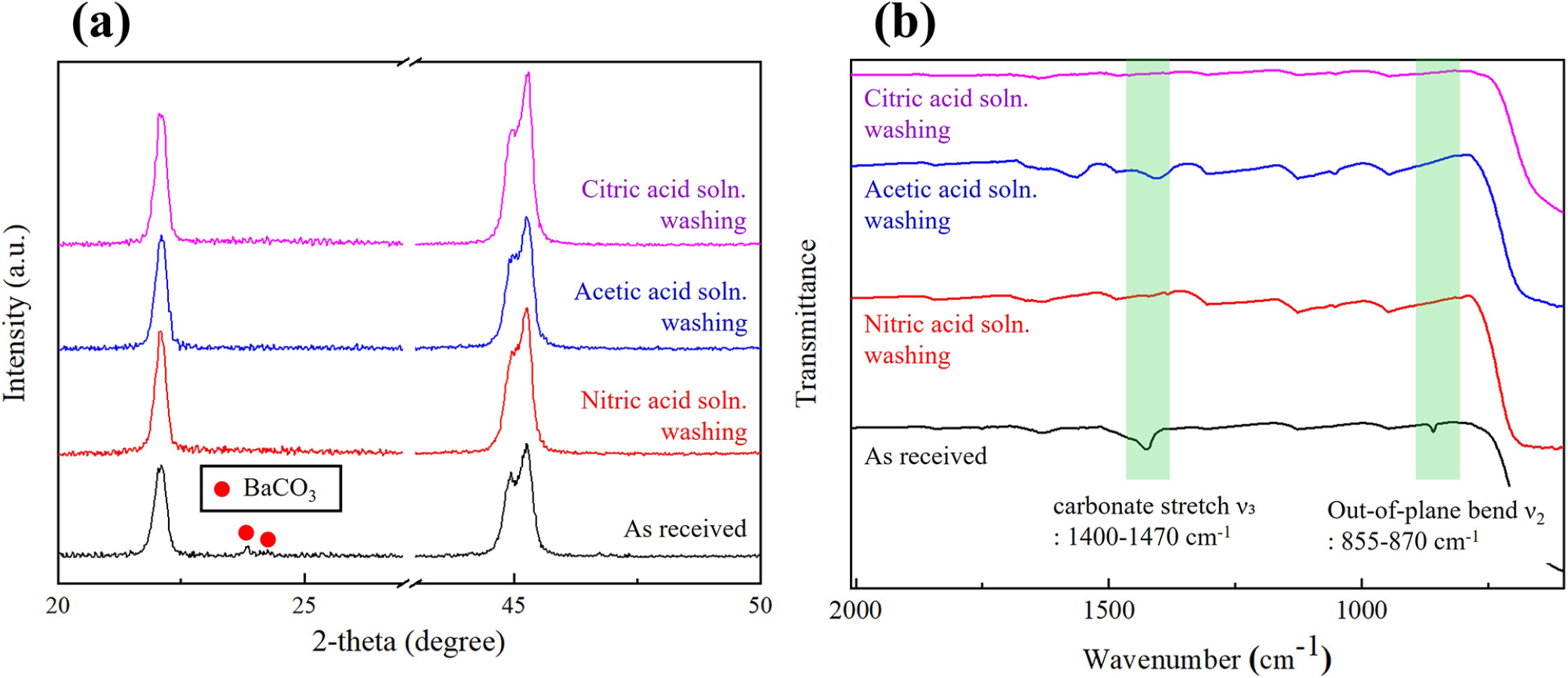
Fig. 4 demonstrates that residual carbonate associated with hydrothermally synthesized BaTiO3 can be effectively removed by brief acid washing without degrading the perovskite lattice. As shown in Fig. 4(a), the weak reflections attributable to BaCO3 visible in the untreated powder diminish or disappear after washing with nitric, acetic, or citric acid solutions, while the BaTiO3 reflections—especially the (002)/(200) doublet near 2θ ≈ 45°—remain unchanged in position and splitting. Within the precision of our scans, no loss of tetragonality is observed especially for the weak-acid washing, indicating that carbonate removal proceeds without measurable Ba depletion from the perovskite lattice. This is consistent with the cross-checks by the Fourier-transform infrared (FT-IR) spectroscopy analysis. In the FT-IR spectra [Fig. 4(b)], the as-received powder shows characteristic BaCO3 bands—most prominently the asymmetric carbonate stretch ν3 near 1,410-1,470 cm-1 and the out-of-plane bend ν2 near ~855-870 cm-1—superimposed on the BT framework modes. After acid washing, these carbonate bands are strongly suppressed; however, small residual ν3 features remain detectable for the nitric- and acetic-acid-washed powders, whereas they are completely removed to the noise level after citric-acid washing. In all three cases the BT lattice bands are essentially unchanged, indicating selective removal of surface and intergranular carbonate, but the FT-IR results highlight citric acid as the most efficient carbonate-cleaning agent among the three. Mechanistically, mild acids protonate surface CO32-/HCO3- species and dissolve BaCO3 as soluble Ba2+ salts; citrate can further assist via weak chelation of Ba2+, which likely contributes to the more complete desorption of carbonate from the BT surface. These outcomes are consistent with reports that carbonate formed under highly alkaline hydrothermal conditions is readily removed by controlled acidification, provided exposure is short and pH is carefully neutralized afterward.19,31) From a synthesis-property standpoint, these observations close the loop suggested in Figs. 1, 2, 3: carbonate formed more readily under Ba-rich, strongly alkaline conditions can lower effective tetragonality by locally scavenging Ba2+ during growth; its post-synthetic removal restores stoichiometry at interfaces and helps preserve the tetragonal signatures established at the optimal Ba/Ti ≈ 1.7. Practically, we find that 0.5-1.0 M citric acid affords nearly complete suppression of carbonate bands while maintaining the BT lattice, with 0.5-1.0 M acetic acid also providing substantial—though slightly less complete—carbonate removal; both are preferable to strong oxidizing HNO3 from a process-safety and waste-handling perspective. Taken together, Fig. 4 supports a simple and safe “carbonate-clean” protocol, with citric-acid washing in particular offering an effective and scalable route to maintain the tetragonal BaTiO3 structure confirmed by XRD and FT-IR in this study.
4. Conclusion
This study establishes a carbonate-suppressed hydrothermal route for synthesizing highly tetragonal BaTiO3 (BT) nanoparticles directly from commercial TiO2 (P25) and barium acetate in KOH media at 240 °C for 6 h, without any post-annealing. Relative to the conventional Ba(OH)2 route, the acetate-based process markedly suppresses BaCO3 formation, yields finer and narrower particle-size distributions (D50 ≈ 85 nm), and produces powders that are already tetragonal at room temperature, as confirmed by clear (002)/(200) XRD peak splitting and Raman fingerprints of the ferroelectric phase.
Systematic control of the Ba/Ti ratio revealed an optimal composition window centered at Ba/Ti = 1.7, where BT exhibits the highest tetragonality (c/a = 1.007615). Below this ratio, residual Ba deficiency during hydrothermal nucleation and growth likely limits the tetragonal distortion, while excessive Ba above this range promotes carbonate formation that scavenges Ba2+ and subtly degrades effective tetragonality despite an apparent Ba surplus. These findings provide practical guidelines for balancing Ba excess and carbonate suppression to maximize ferroelectric symmetry in hydrothermal BT.
Post-synthetic weak-acid washing, particularly with 0.5-1.0 M citric acid, efficiently removes trace carbonate while preserving the BT lattice, as evidenced by the disappearance of BaCO3 FT-IR bands and the unchanged perovskite signatures. Compared with strong oxidizing acids, such weak-organic-acid treatments combine high carbonate-removal efficacy with improved safety and simpler waste handling, offering a realistic finishing step for industrial powder processing.
Taken together, the barium-acetate-based hydrothermal process, optimized Ba/Ti chemistry, and mild carbonate-cleaning protocol provide an economical, energy-efficient, and scalable pathway to phase-pure, highly tetragonal BT nanoparticles. The demonstrated control over impurity phases, defect chemistry, and particle-size uniformity directly addresses key requirements for next-generation MLCC feedstock powders and offers a robust platform for future investigations of sintering behavior, dielectric reliability, and doped BT systems tailored to advanced capacitors and electronic applications.
