

1. Introduction
2. Materials and Methods: Computational Investigation
3. Results and Discussion
3.1. Frontier molecular orbitals and electronic properties
3.2. Photovoltaic properties
3.3. Absorption properties
4. Conclusion
1. Introduction
The development of π-conjugated donor-acceptor (D-A) molecular systems have garnered significant attention in the area of organic electronics owing to their interesting properties.1-11) The optoelectronic properties of the π-conjugated D-A molecular systems are likely to depend on the type, number and mode of linkage between the donating and the accepting units.11-14) Apart from the aforementioned reasons tuning of properties via substitution of H-atoms with other atoms or groups is also an effective design strategy in these D-A molecular systems.15-24) The quest for development of π-conjugated D-A molecular systems for organic photovoltaics with improved efficiency still continues as organic photovoltaics is a promising substitute for renewable energy source. In this respects the study of bulk heterojunction (BHJ) solar cells are important owing to their notable advantages that is flexibility and low cost.1,2,8,17) To capture solar energy, organic BHJ solar cells typically include electron-donating and electron-accepting materials (mostly PCBM or one of its derivatives).3,8,16,17) The π-conjugated D-A organic materials are highly explored for BHJ solar cells because of their ease of synthesis, solution-processable materials, low cost, together with high performance.1-3,8,16,17) In the recent past, considerable theoretical and experimental work has been dedicated to study photovoltaic properties in order to improve the PCE (power conversion efficiencies).1,3,5,16,17,22-35) This inspires us to study π-conjugated D-A small organic molecules for organic solar cells (SMOSCs). The efficiency of SMOSCs depends on the light harvesting ability of the π-conjugated D-A small organic molecules which must efficiently absorb at the solar radiation in the visible region. The push-pull structure of the π-conjugated D-A small organic molecules enhances intramolecular charge transfer (ICT), which reduces the HOMO-LUMO gap and enhances excitation charge transfer and transport characteristics.22-26)
In this paper, we report four π-conjugated D-A small molecules derived from triphenylamine (TPA) and benzothiadiazole (BTD) building block. The designed D-A-π-A SMD1-4 are shown in Fig. 1. The theoretical studies and comparison of the results show that functionalization with additional fluorine and cyano groups on the BTD unit can be an effective strategy to regulate the HOMO-LUMO gap and tune the optoelectronic properties of π-conjugated D-A small molecules. We have used the DFT and TD-DFT method to study and analyze the optoelectronic, and photovoltic properties of these π-conjugated D-A small organic molecules. We feel the present study could assist to develop efficient π-conjugated D-A small molecular materials, for BHJ solar cells.
2. Materials and Methods: Computational Investigation
The optimized molecular structures of SMD1-4 and their properties such as HOMO and LUMO levels, and the HOMO-LUMO gap (Eg) are obtained utilizing the DFT/B3LYP/6-31G(d,p) level.22-25,29,33) The TD-DFT calculation using CAM-B3LYP functional and 6-31G(d,p) basis set were performed to investigate the oscillator strengths (f) and the optical transitions.22-25,29,33) All the calculation were performed using Guassion 09 package.36)
3. Results and Discussion
3.1. Frontier molecular orbitals and electronic properties
The theoretical information about the HOMO and LUMO energy levels of D-A molecular systems plays a crucial role in exploring organic photovoltaic properties.26,29) The effectiveness of the charge transfer is crucial for photovoltaic devices and depends on the HOMO and LUMO energy levels of donor and acceptor units.26) DFT calculations can be employed to investigate the HOMO and LUMO energies.29-33)
The frontier molecular orbitals (FMO) distribution pattern and the calculated values for HOMO, LUMO and HOMO-LUMO gap for D-A-π-A SMD1-4 at B3LYP/6-31G(d,p) level is shown in Fig. 2. The HOMOs for all the small molecule donors are mainly delocalized on the triphenylamine moiety and the benzo part of the adjacent BTD unit. The LUMOs are mainly delocalized on two BTD core. This indicates that TPA act as a donor, and the BTD groups act as an acceptor. A closer look at the FMO distribution pattern shows that in SMD1 and SMD4 the delocalization of LUMO is equally over both the acceptors. On contrary in SMD2 and SMD3 the delocalization of LUMO is more on the cyano substituted benzothiadiaole unit. This indicates stronger electron withdrawing nature of cyano substituted benzothiadiaole unit.14,18,19,21)
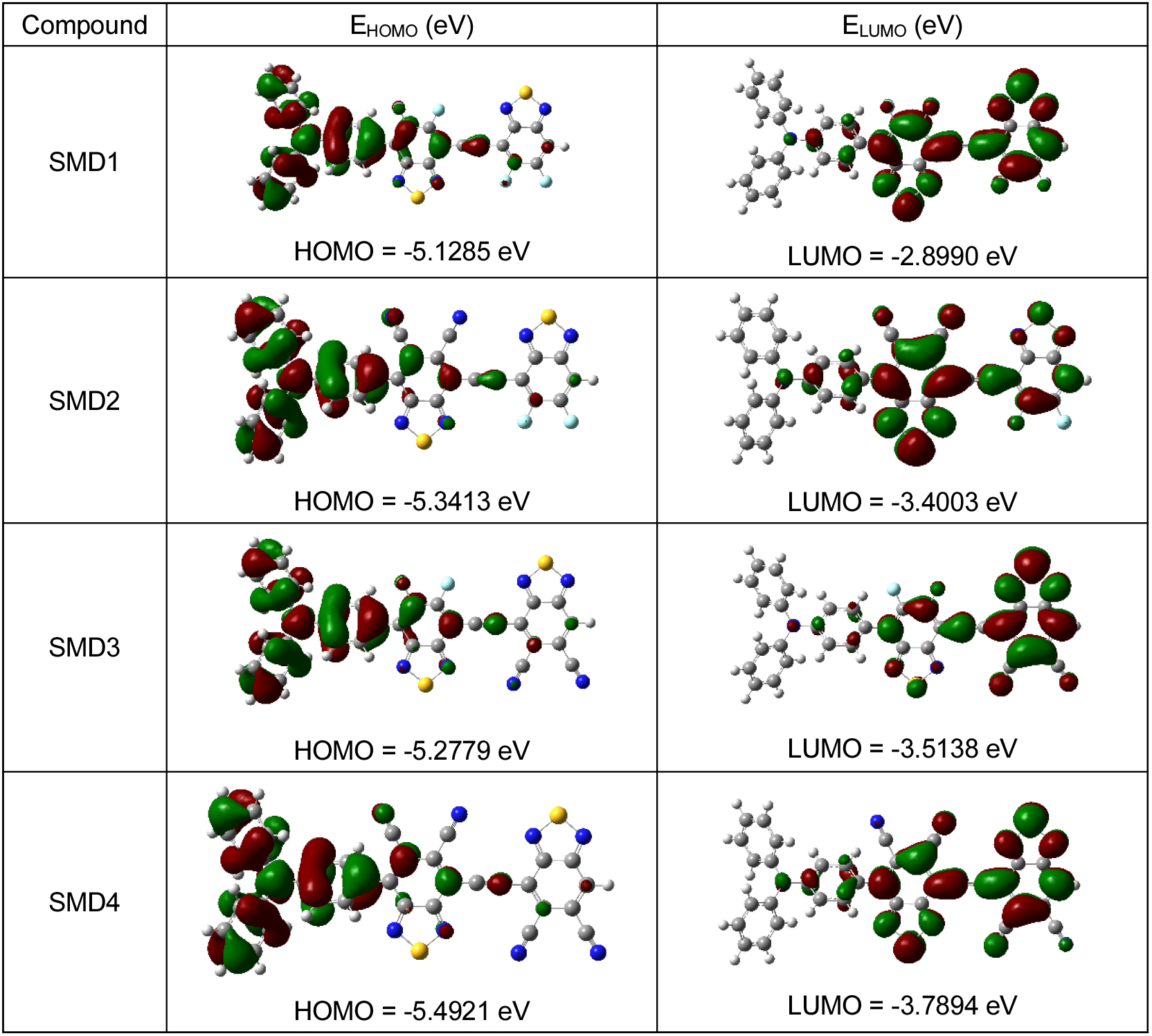
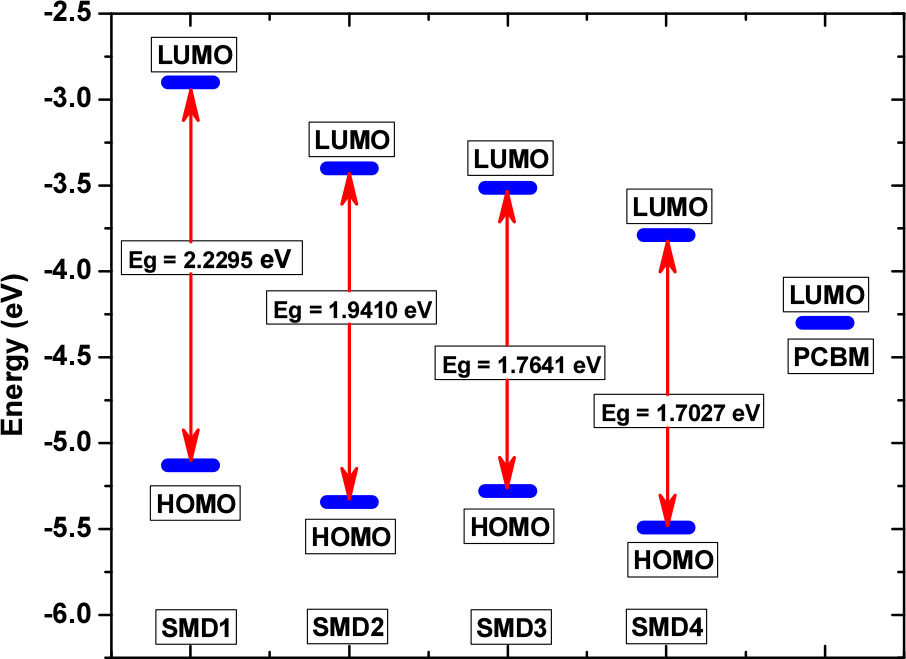
The values of HOMO energies for SMD1-4 are -5.1285 eV, -5.3413 eV, -5.2779 eV and -5.4921 eV respectively. The HOMOs of SMD1-4 follows the trend SMD1 > SMD3 > SMD2 > SMD4. The values of LUMO energies for SMD1-4 are -2.8990 eV, -3.4003 eV, -3.5138 eV and -3.7894 eV respectively. The LUMOs of SM1-4 follows the trend SMD1 > SMD2 > SMD3 > SMD4. The values of energy gaps for SMD1-4 are 2.2295 eV, 1.9410 eV, 1.7641 eV and 1.7027 respectively. The calculated HOMO-LUMO gap (Eg) of the studied model compounds follow the trend SMD1 > SMD2 > SMD3 > SMD4. The decrease in the Eg values for SMD1-4 (Fig. 3) can be explained considering two factors: (a) The presence of stonger electron withdrawing groups and (b) The dihedral angle between the donor triphenyamine and the acceptor BTD units. SMD4 with two strongly withdrawing cyno substituted BTDs exhibits lowest Eg value. The trend indicates stronger electron accepting character of cyano group substituted BTD as compared to fluorine group substituted BTD.3) The presence of fluorine groups on the BTD unit placed adjacent to the triphenylamine unit results in smaller dihedral angle (33.969°) in case of SMD3 as compared to SMD2 (43.022°) with cyano substituted BTD unit. This results larger planarity, stronger donor-acceptor interaction and lower HOMO-LUMO gap for BTD3 as compared to SMD2.3) The trend indicates stronger electron accepting character of cyano group substituted BTD as compared to fluorine group substituted BTD.19,21,37-39)
3.2. Photovoltaic properties
In general power conversion efficiency (PCE) of solar cell is a significant parameter to compare their performance. The PCE depends on JSC (short-circuit current density), Voc (open circuit voltage), FF (fill factor), and the Pinc (incident photon to current efficiency) and can be calculated according to the expression given below:29,33)
The open-circuit voltage (Voc) of BHJ is determined from the difference between the HOMO of the donor (π-conjugated molecule) and LUMO of the acceptor, taking into account the energy lost during the photo-charge generation.29) The theoretical values of open-circuit voltage Voc of the BHJ solar cell have been calculated from the following expression:17,24)
The theoretic values of Voc for the studied molecular system SMD1-4 are 0.5285 eV, 0.7413 eV, 0.6779 eV, 0.8921 eV in the case of PC71BM (Table 1). The Voc value follows the order SMD4 > SMD2 > SMD3 > SMD1. The higher Voc value for SMD4 and SMD2 as compared to SMD1 and SMD3 in these D-A-π-A molecular system is attributed to the deepened HOMO level of SMD2 and SMD4. The comparison of Voc values reveals that substitution of fluorine and cyano group on the BTD core results in significant variation in the Voc values and can be utilized as effective strategy to develop molecular systems with improved PCE.19,21)
Table 1.
Energy values of ELUMO (eV), EHOMO (eV), Egap (eV) and the open circuit voltage Voc (eV) and LD - LA of the studied molecules obtained by B3LYP/6-31G(d,p) level.
Table 1 shows (LD - LA) the difference between of LUMO energy levels between those new designed donors (SMD1-4) and the PC71BM acceptors. These calculated values of (LD - LA) are appropriate for efficient electron injection from the donor to the LUMO of the PC71BM acceptor Based on the theoretical results, both the studied molecules can be used as efficient donors for BHJ solar cells.29)
The FF (fill factor) parameter valued for SMD1-4 were calculated and compiled in Table 1.5) The FF value follows the order SMD4 > SMD2 > SMD3 > SMD1. The results shows that the FF parameter can be tuned by altering the fluorine and cyano substituents for improving PCE.
The prediction of PCE for organic solar cells based on Scharber diagram have been widely used.17) The theoretical PCE of the designed molecules SMD1-4 is between ~1.5 % to ~7.5 % from initial assessment from Scharber diagram.17,19,29) The SMD4 exhibits the highest PCE value among SMD1-4. The results illustrate that substitution of cyano group can improve the solar cell efficiency more than the fluorine substitution.
3.3. Absorption properties
The TD-DFT computations at CAM-B3LYP has become an important tool to explore the optical properties of π-conjugated molecular systems.24,29,33) Therefore in order to study the electronic transitions for SMD1-4, the quantum calculation was performed using TD-DFT/CAMB3LYP/6-31G(d,p) level.29,33) The calculated absorption wavelengths (λmax), oscillator strengths (ƒ) and vertical excitation energies (E) for solvent (chloroform) were carried out and the data is compiled in Table 2.
Table 2.
Calculated electronic absorption spectra for SMD1-4.
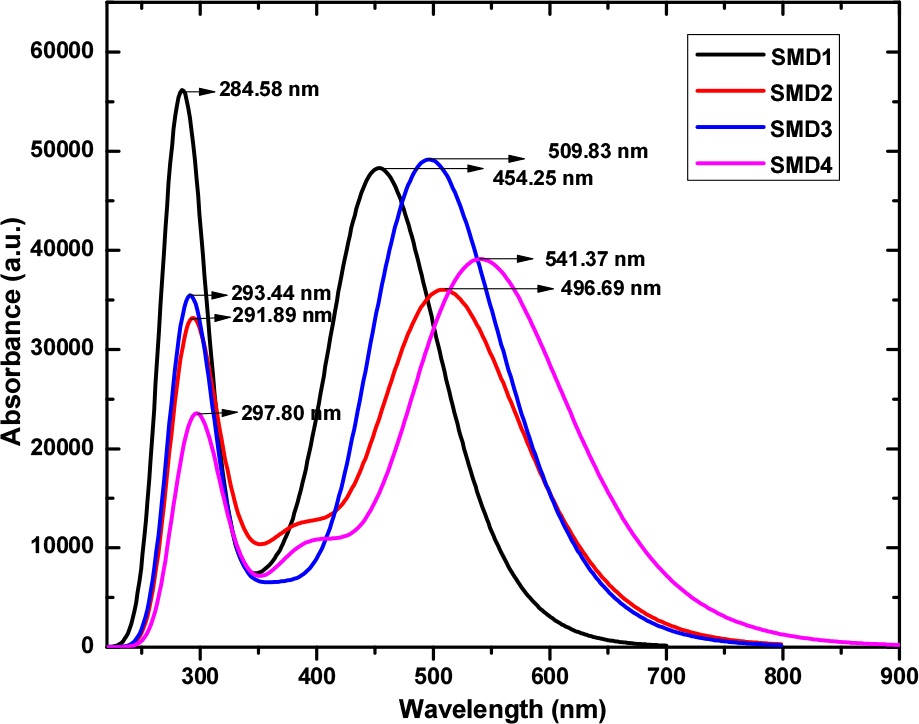
The UV-vis spectra for SMD1-4 exhibit a similar absorption profile which shows two intense bands in chloroform solution (Fig. 4).14) The higher energy band for SMD1-4 attributed to the π→π* transition was observed in the region of 280 nm to 300 nm and was not strongly influenced by structural variation. On the contrary the lower energy transition for SMD1-4 due to intramolecular charge transfer (ICT) was observed in the region of 450 nm to 550 nm and was strongly influenced by the structural variation induced due fluorine and cyano group substitution on the BTD core.40,41) The UV-vis absorption shows a large bathochromic shift (~87 nm) for SMD4 as compared to SMD1 when the fluorine substituted BTD acceptor units in D-A-π-A architecture were replaced by strongly electron withdrawing cyano substituted BTD acceptor unit. However SMD1 and SMD3 with fluorine substituted BTDs adjacent to the triphenylamine unit exhibit higher absorption coefficient compared to SMD2 and SMD4. This can be accounted to greater planarity, and extension of the π-conjugation between the donor triphenyamine and the adjacent fluorine substituted BTD in SMD1 and SMD3 compared to SMD2 and SMD4 with adjacent cyano substituted BTD.42) The TD-DFT calculation results clearly indicate that significant tuning of the optical absorption can be achieved by fluorine and cyno group substitution in the D-A-π-A BTDs.19,37-39)
4. Conclusion
In summary, a series of D-A-π-A system SMD1-4 featuring triphenylamine donor and benzothidiazole acceptor have been designed. The molecular design hinges on the incorporation of cyano and fluorine groups on the benzothiadizole unit. The DFT and TDDFT methods were used to study the optoelectronic properties, calculation of HOMO, LUMO and HOMO-LUMO gap. The functionalization of these D-A-π-A systems with cyano and fluorine groups significantly perturbs the FMOs and tuning of the HOMO-LUMO gap. The open-circuit voltage for SMD1-4 with PCBM acceptor, clearly shows the impact of cyano and fluorine group substitution in these D-A-π-A systems. The findings herein will be interesting for development of cyano and fluorine functionalized D-A systems for photovoltaic applications.
